

Fabrication of magnetic Fe3O4@SiO2@TiO2-Au with core-shell structure and its photocatalytic reduction activity
-
摘要: TiO2基的光催化剂已被广泛用于各种有机物污染物的光氧化和水中六价铬Cr(VI)的光还原。然而,对光催化还原硝基芳香化合物为胺基芳香化合物的研究鲜有报道。本文采用液相沉积(LPD)法将锐钛矿型TiO2沉积在非晶SiO2包覆的Fe3O4上,制备了核壳结构的Fe3O4@SiO2@TiO2磁性光催化剂。为进一步提高其光催化活性,将均匀分散的Au纳米粒子(Au NPs)修饰在其表面,以获得Fe3O4@SiO2@TiO2-Au纳米复合材料。对这两种TiO2基的磁性复合材料进行表征并将其用作光催化剂。在紫外光照射下,用HCOONH4作为空穴捕集剂和H源,以对硝基苯胺(p-NA)光催化还原至对苯二胺(p-PDA)作为模型反应,评价其光催化还原性能。结果表明:虽然两种光催化剂都能将p-NA完全还原成p-PDA,但Fe3O4@SiO2@TiO2-Au表现出比Fe3O4@SiO2@TiO2更优异的光催化活性。这是由于TiO2表面修饰的Au NPs能有效地促进光激发的电子从TiO2的导带转移到Au,最大限度地减少电子和空穴的复合率,延长光电子的寿命。此外,不可或缺的HCOONH4在p-NA的光催化还原中能有效地捕获光生空穴,极大地提高了其光催化还原效率。Abstract: TiO2-based photocatalysts have been widely used for photoxidation of various organic pollutants and photoreduction of Cr(VI) in aqueous solution. However, less attention has been paid to the photocatalyzed reduction of nitro group to amino group of nitroaromatics. In this study, liquid-phase deposition (LPD) technique was adopted to deposit anatase TiO2 shell on amorphous SiO2-coated Fe3O4 to form core-shell Fe3O4@SiO2@TiO2 magnetic photocatalyst. To further promote its photocatalytic activity, uniformly dispersed Au nanoparticles (NPs) were decorated on its surface to obtain Fe3O4@SiO2@TiO2-Au composite. Both the TiO2-based magnetic composites were characterized and used as photocatalysts. To evaluate their photocatalytic performance, the photocatalyzed reduction of p-nitroaniline (p-NA) to p-phenylenediamine (p-PDA) was chosen as a model reaction under UV irradiation using HCOONH4 as a hole scavenger and H source. Although p-NA can be completely reduced to p-PDA by the two photocatalysts, Fe3O4@SiO2@TiO2-Au exhibits much superior photocatalytic activity to Fe3O4@SiO2@TiO2. The is because Au NPs decorated on the TiO2 can efficiently facilitate the photoexcited electron-transfer from the conduction band of TiO2 to Au, which is favorable for minimizing the recombination rate of electrons and holes and prolong the lifetime of the photoelectrons. In addition, HCOONH4 plays an indispensable role in improving the photocatalytic reduction of p-NA via capturing photo-generated holes.
-
Keywords:
- magnetism /
- TiO2 /
- core-shell structure /
- Fe3O4@SiO2@TiO2-Au /
- photocatalytic reduction /
- p-nitroaniline
-
硝基芳香化合物,如硝基苯(NB)、对硝基苯酚(p-NP)、对硝基苯胺(p-NA) 和Cl或Br取代的硝基苯(Cl-NB或Br-NB)等,已被广泛地用作合成各种精细化学品的前体或中间体。然而,工业废水中排放的硝基芳烃污染物即使在低浓度下也有很强的毒性和致突变性,并且由于其危害性,已被列为需优先处理的污染物[1-5]。目前,一些水处理技术如物理吸附、反渗透、光催化氧化及生物方法等从废水中去除这些污染物[2-5]。然而这些方法处理不彻底,甚至会造成二次污染,其次处理后的产物附加值较低,回收难,因此迫切需要开发一种对环境无害的催化工艺,将有毒的硝基芳烃转化为毒性更低、附加值更高、且易回收的氨基衍生物。到目前为止,主要是采用热催化还原法,即用各种贵金属或它们的合金纳米粒子作为催化剂,过量的NaBH4、水合肼或氢硅烷作为还原剂和氢源将硝基催化还原为氨基[6-9]。然而,额外的还原剂依然对环境造成严重污染。
在过去的几十年里,TiO2基的光催化作为一种潜在的解决环境污染的方法引起了极大的关注。在紫外光照射下,TiO2价带和导带产生的光生空穴(h+)和电子(e−)分别具有很强的氧化和还原能力,现有光化学或光催化的研究主要集中在利用其价带(VB)产生的空穴(h+)光催化氧化各种有机污染物[10];导带(VB)上光生电子(e−)将高毒性的六价铬Cr(VI)还原成低毒性的三价铬Cr(III)或将H+还原为H2[11-12]。而对光催化剂产生的光电子直接将硝基芳香化合物光还原为氨基芳香化合物的研究鲜有报道。另一方面,作为光催化剂,单组分纳米TiO2存在两个固有缺陷[13]:(1) 光生电子(e−)和空穴(h+)易发生复合,光催化效率较低;(2) 光催化反应后难以分离和循环使用,可能造成二次污染。
将TiO2与Au、Ag、Pt和Pd等贵金属复合,复合光催化剂中有效成分之间的协同作用可以极大地促进其光催化活性[14-15]。而磁性材料具有易于分离、回收和循环利用的优势,被广泛应用于废水处理和催化剂载体等[16-18]。Jin等[19]制备了 Fe3O4@TiO2-Au催化剂并用于降解2, 4-二硝基苯,同时用表面增强拉曼光谱监测催化过程。Li等[20]通过溶胶-凝胶工艺在Fe3O4上涂覆一层 TiO2,然后通过水热处理使 TiO2 晶化并形成介孔,再通过高氯酸原位还原法在 Fe3O4@mTiO2微球上沉积金纳米粒子,该催化剂表现出良好的光催化性能。Chi等[21]制备了Fe3O4@SiO2@TiO2-Ag 催化剂,且其催化活性高于商用TiO2 (P25)。Li等[22]制备了Fe3O4@SiO2@TiO2-Pt磁性催化剂,其TiO2为有序分层结构,并暴露较多(001)晶面,结合Pt纳米粒子后表现出较好的光降解性能。然而,先前的研究所用钛氧有机化合物水解速率太快,生成的TiO2易团聚,难以获得理想的核壳结构[23]。此外,通过溶胶-凝胶沉积的非晶TiO2需要煅烧才能转变成晶型TiO2,煅烧能将磁性Fe3O4氧化为非磁性的α-Fe2O3[24]。本工作用层层组装战略制备Fe3O4@SiO2@TiO2-Au核壳结构的纳米复合光催化剂:首先,结合溶胶-凝胶法和液相沉积(LPD)法制备超顺磁性核壳结构Fe3O4@SiO2@TiO2纳米微球。然后,通过种子介导生长法,在其表面负载Au纳米粒子(NPs)。与现有方法相比,新颖性主要包括:(i) 在室温和水介质中将锐钛矿型TiO2 NPs均匀地沉积在Fe3O4@SiO2表面,厚度可控;(ii) 种子介导生长法在TiO2上负载均匀分布的Au NPs提高光催化性能;(iii) Fe3O4@SiO2@TiO2-Au作为光催化剂能将p-NA完全还原成对苯二胺(p-PDA),不需要外加还原剂,符合绿色化学的基本原则[25]。
1. 实验方法
1.1 原材料
实验使用的所有化学试剂均为分析级试剂:六水三氯化铁(FeCl3·6H2O)、六氟钛酸铵((NH4)2TiF6)、硼酸(H3BO3)、氨水(NH3·H2O,25%~28%)、尿素、甲酸铵(HCOONH4)、正硅酸乙酯、乙二醇(EG)、柠檬酸钠(Na3Cit)、硼氢化钠(NaBH4)、对硝基苯胺(p-NA)和对苯二胺(p-PDA)由国药集团化学试剂有限公司生产;氯金酸和聚二甲基二烯丙基氯化铵(PDADMAC,重均分子量Mw <
100000 ,35%水溶液)由默克试剂公司生产。1.2 核壳结构的制备
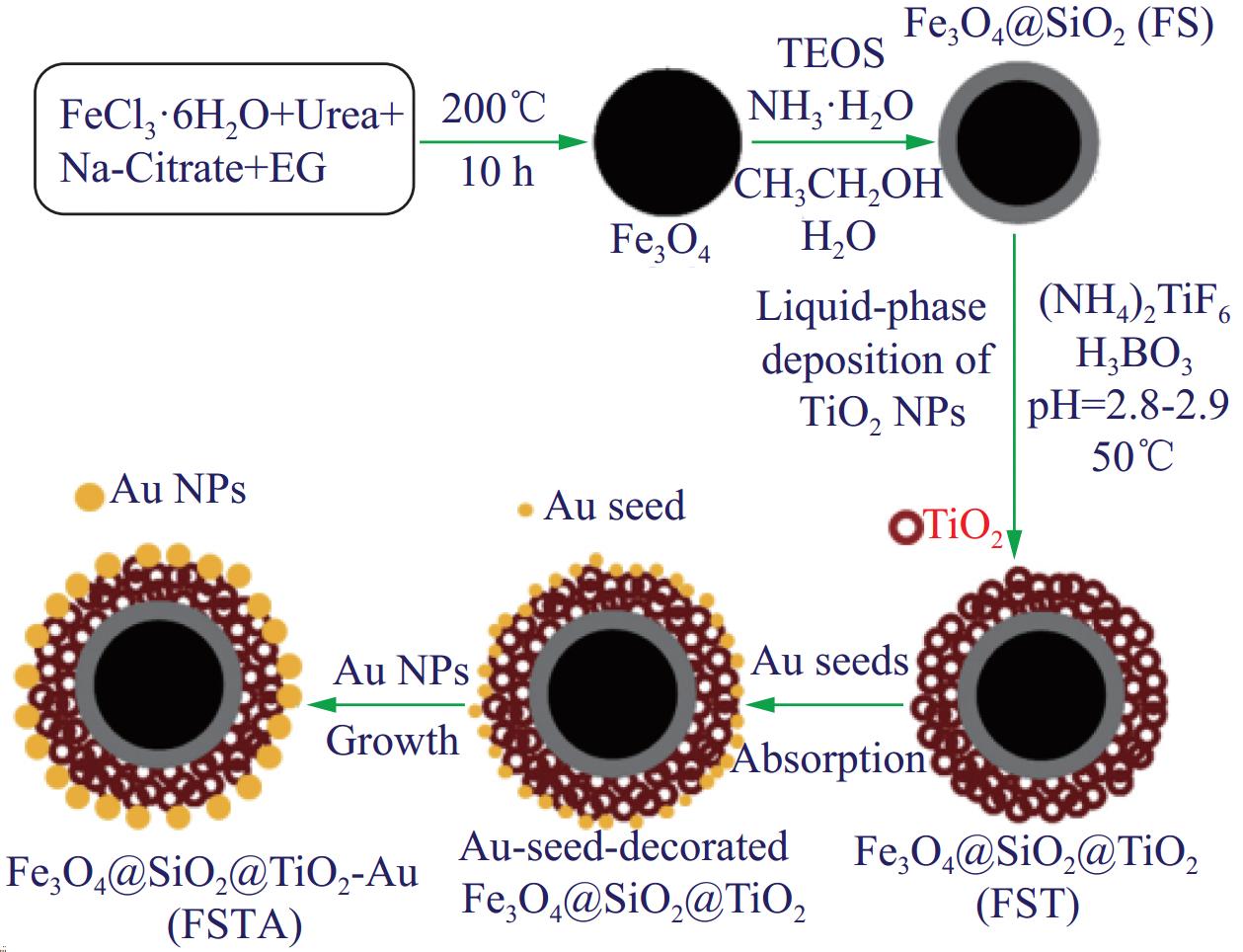
多层核壳结构Fe3O4@SiO2@TiO2-Au的制备流程如图1 所示,主要包括下列过程:溶剂热法合成Fe3O4微球、sol-gel法进行中间层SiO2包覆、LPD法在SiO2表面沉积外层TiO2及阳离子聚合物对TiO2表面改性和Au NPs的表面修饰。
![]() 图 1 磁性Fe3O4@SiO2@TiO2-Au核壳结构微球的组装流程图Figure 1. Schematic diagram for fabricating magnetic Fe3O4@SiO2@TiO2-Au microspheres with core-shell structureNPs—Nanoparticles; EG—Ethylene glycol; TEOS—Tetraethyl orthosilicate
图 1 磁性Fe3O4@SiO2@TiO2-Au核壳结构微球的组装流程图Figure 1. Schematic diagram for fabricating magnetic Fe3O4@SiO2@TiO2-Au microspheres with core-shell structureNPs—Nanoparticles; EG—Ethylene glycol; TEOS—Tetraethyl orthosilicate1.2.1 Fe3O4@SiO2@TiO2制备及其表征
按照文献方法制备单分散及羧基功能化的超顺磁性Fe3O4微球[26]。通过我们以前报道的方法制备核壳结构的Fe3O4@SiO2和Fe3O4@SiO2@TiO2 [27]。
Fe3O4@SiO2@TiO2静电吸附阳离子聚合物PDADMAC对其进行表面改性:将0.1 g Fe3O4@SiO2@TiO2超声分散到150 mL含有50 mg PDADMAC的水溶液中,搅拌6 h然后静置2 h达到吸附平衡。磁分离PDADMAC修饰的Fe3O4@SiO2@TiO2微球,并用去离子水洗涤3次以去除表面未牢固吸附的聚合物。
1.2.2 在改性的Fe3O4@SiO2@TiO2表面锚固Au晶种
在Na3Cit溶液中,通过NaBH4还原HAuCl4制备粒径尺寸约为3 nm的Au溶胶[28]。将0.1 g PDADMAC修饰的Fe3O4@SiO2@TiO2分散到50 mL该Au溶胶中,通过静电吸附将Au晶种锚固在其表面。静置2 h后,通过磁分离,蒸馏水洗涤,去除未被吸附的Au溶胶。
1.2.3 种子生长法在Fe3O4@SiO2@TiO2上生长金纳米粒子
将吸附了Au晶种的Fe3O4@SiO2@TiO2分散到100 mL含有25 mg K2CO3和2 mL 0.1% HAuCl4的镀金溶液中,加入0.5 mL甲醛溶液以还原Au3+并在TiO2表面吸附的Au晶种上均匀生长Au NPs,通过磁分离和洗涤得到Fe3O4@SiO2@TiO2-Au纳米复合光催化剂。
1.3 材料表征
通过透射电镜 (Tecnai G2 F30,美国FEI)对样品进行普通形貌(TEM)、高分辨透射电镜图像(HRTEM)、高角度环形暗场扫描透射电子显微镜图像(HAADF-STEM)和元素分布图像(element mapping)进行分析;用X射线粉末衍射仪(XRD,德国布鲁克D8)以Cu Kα为辐射源检测样品的晶体结构,扫描范围为2θ=5°~80°;以Al Kα为辐射源;通过X射线光电子能谱(XPS,PHI-5702,美国)分析样品的组成和价态;使用磁学测量系统(SQUID MPMS C-151,Quantum Design,美国)测量样品磁性。
1.4 光催化还原性能评价
将50 mg Fe3O4@SiO2@TiO2或Fe3O4@SiO2@TiO2-Au催化剂超声分散到250 mL的p-NA溶液(20 mg·L−1)中,加入200 mg HCOONH4,将溶液转入光催化反应器中[29]。光照之前,上述悬浮液在黑暗中搅拌30 min,以达到吸附平衡;然后在298 K、高压汞灯(500 W、λmax=365 nm)进行光催化反应。在反应过程中,以设定的时间间隔吸取5 mL样品溶液,磁分离以去除催化剂,在紫外-可见分光光度计(UV-2600 型,日本 Shimadzu 公司)上测量该溶液的紫外-可见(UV-Vis)吸收光谱。对照实验在3种不同的条件下进行:(1) p-NA+HCOONH4不加化剂;(2) p-NA+HCOONH4+光催化剂无光照射;(3) p-NA+光催化剂不加HCOONH4。光催化过程始终通入氮气保护。
2. 结果与讨论
2.1 Fe3O4@SiO2@TiO2-Au的微观形貌
在尿素和Na3Cit的存在下,通过EG高温还原Fe(III)盐而制备出磁性Fe3O4亚微米球[26]。 Na3Cit的3个羧基对Fe3+离子有很强的配位亲和力,作为稳定剂,尿素作为均匀沉淀剂,可以促进Fe3O4在多元醇溶剂中的形成。图2为Fe3O4的TEM图像。从图中可以看出,Fe3O4呈现出球形结构,单分散性好,其平均直径约为380 nm (图2(a))。 图2(b)表明,Fe3O4亚微米球实际上是由许多尺寸小于10 nm的小纳米晶构成。 通过TEOS的水解和缩合的sol-gel过程,在Fe3O4亚微米球表面形成均匀光滑的SiO2壳层 (厚度约30 nm),从而形成核壳Fe3O4@SiO2亚微米球 (图2(c)和图2(d))。沉积在Fe3O4上的SiO2表面由于存在硅羟基(Si—OH),在较大的pH值范围内带负电荷,通过静电吸附一层阳离子PDADMAC聚电解质后,使Fe3O4@SiO2表面带正电荷。在 LPD过程中,反应介质中带负电荷的[TiF6]2−阴离子吸附在带正电的Fe3O4@SiO2表面,原位水解而直接转化为锐钛型TiO2 NPs,形成Fe3O4@SiO2@TiO2。H3BO3用于清除水解和转化过程中产生的F−离子,促使水解平衡向右移动[27, 30]。图2(e)和图2(f)为Fe3O4@SiO2@TiO2在不同放大倍数下的TEM图像。可以清楚地看到,Fe3O4@SiO2表面沉积了一层致密均匀的TiO2 NPs外壳。由于在光催化反应中,Fe3O4能成为光生空穴(h+)-电子(e−)的结合中心,捕获TiO2产生的光电子,降低光催化效率[24]。因此为了避免Fe3O4不利影响,在Fe3O4和TiO2之间插入SiO2夹层,构建核壳型Fe3O4@SiO2@TiO2。经PDADMAC功能化的Fe3O4@SiO2@TiO2表面带正电,因此带负电的Au晶种可以很容易地被吸附在其表面 (图2(g))。这些粒子可以作为晶核,通过甲醛对Au3+的还原,进一步生长出均匀分散的Au NPs。从图2(h)和图2(i)中可以看出,种子介导生长制备的单分散的Au (平均粒径5.2 nm)牢固地负载在TiO2的表面,所形成核壳结构的Fe3O4@SiO2@TiO2-Au复合球是由SiO2包覆的Fe3O4核心、有序的TiO2外壳和分散在TiO2表面的Au 纳米粒子组成。图2(j)为TiO2和Au NPs的HRTEM图像,清楚地表明了它们的晶体结构特征。测得的晶格间距约为0.351 nm和0.236 nm,分别对应锐钛矿TiO2的(101)和Au NPs的(111) 晶面[15-17]。
![]() 图 2 Fe3O4 ((a), (b))、Fe3O4@SiO2 ((c), (d))、Fe3O4@SiO2@TiO2 ((e), (f))、Au-seed-decorated Fe3O4@SiO2@TiO2 (g) 和 Fe3O4@SiO2@TiO2-Au ((h), (i))的不同放大倍数的TEM图像;(j) 沉积在Fe3O4@SiO2上的TiO2和Au纳米粒子(NPs)的高分辨透射电镜图像Figure 2. TEM images with different magnifications of Fe3O4 ((a), (b)), Fe3O4@SiO2 ((c), (d)), Fe3O4@SiO2@TiO2 ((e), (f)), Au-seed-decorated Fe3O4@SiO2@TiO2 (g), Fe3O4@SiO2@TiO2-Au ((h), (i)); (j) HRTEM of the deposited TiO2 and Au nanoparticles (NPs) on Fe3O4@SiO2
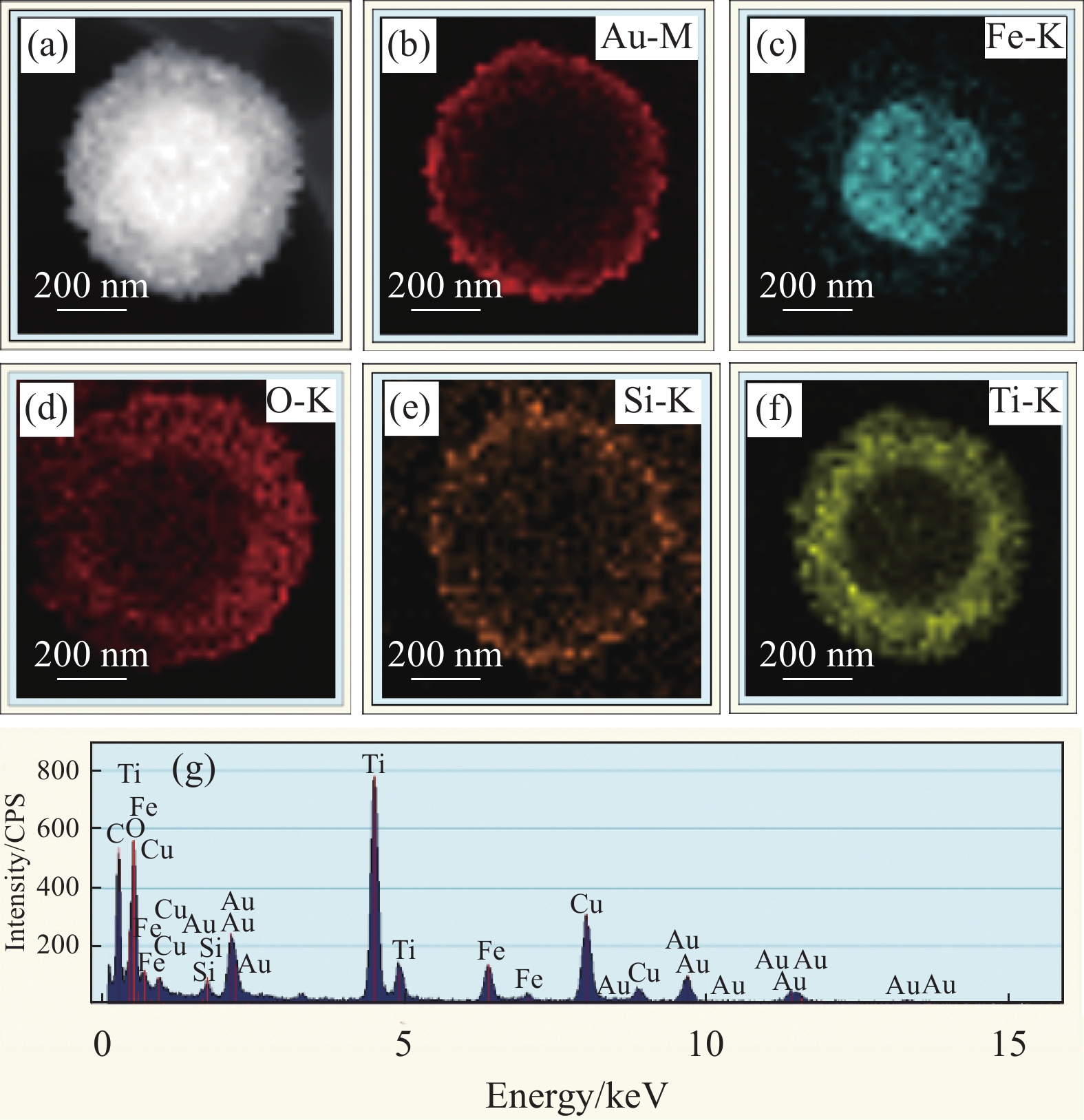
图 2 Fe3O4 ((a), (b))、Fe3O4@SiO2 ((c), (d))、Fe3O4@SiO2@TiO2 ((e), (f))、Au-seed-decorated Fe3O4@SiO2@TiO2 (g) 和 Fe3O4@SiO2@TiO2-Au ((h), (i))的不同放大倍数的TEM图像;(j) 沉积在Fe3O4@SiO2上的TiO2和Au纳米粒子(NPs)的高分辨透射电镜图像Figure 2. TEM images with different magnifications of Fe3O4 ((a), (b)), Fe3O4@SiO2 ((c), (d)), Fe3O4@SiO2@TiO2 ((e), (f)), Au-seed-decorated Fe3O4@SiO2@TiO2 (g), Fe3O4@SiO2@TiO2-Au ((h), (i)); (j) HRTEM of the deposited TiO2 and Au nanoparticles (NPs) on Fe3O4@SiO2图3为Fe3O4@SiO2@TiO2-Au单个复合微球的高角度环形暗场扫描透射电子显微镜(HAADF-STEM)图像、一系列元素分布图及其EDX图谱。图3(g)中的峰对应Fe3O4@SiO2@TiO2-Au中的Fe、O、Si、Ti和Au元素,Cu元素峰来自于铜网。STEM和EDX图谱得到的结果与TEM图像一致,进一步证实了Au NPs被均匀地修饰在Fe3O4@SiO2@TiO2的表面,成功制备了具有核壳结构特征的Fe3O4@SiO2@TiO2-Au纳米复合光催化剂。
![]() 图 3 Fe3O4@SiO2@TiO2-Au复合物的STEM图像(a)、元素谱分布图((b)~(f))、样品的能量色散谱(g)Figure 3. STEM image (a), a series of EDS elemental mapping images (Fe, Si, Ti, O and Au) ((b)-(f)), its corresponding EDX spectrum (g) of Fe3O4@SiO2@TiO2-Au composite
图 3 Fe3O4@SiO2@TiO2-Au复合物的STEM图像(a)、元素谱分布图((b)~(f))、样品的能量色散谱(g)Figure 3. STEM image (a), a series of EDS elemental mapping images (Fe, Si, Ti, O and Au) ((b)-(f)), its corresponding EDX spectrum (g) of Fe3O4@SiO2@TiO2-Au composite2.2 Fe3O4@SiO2@TiO2-Au的晶体结构
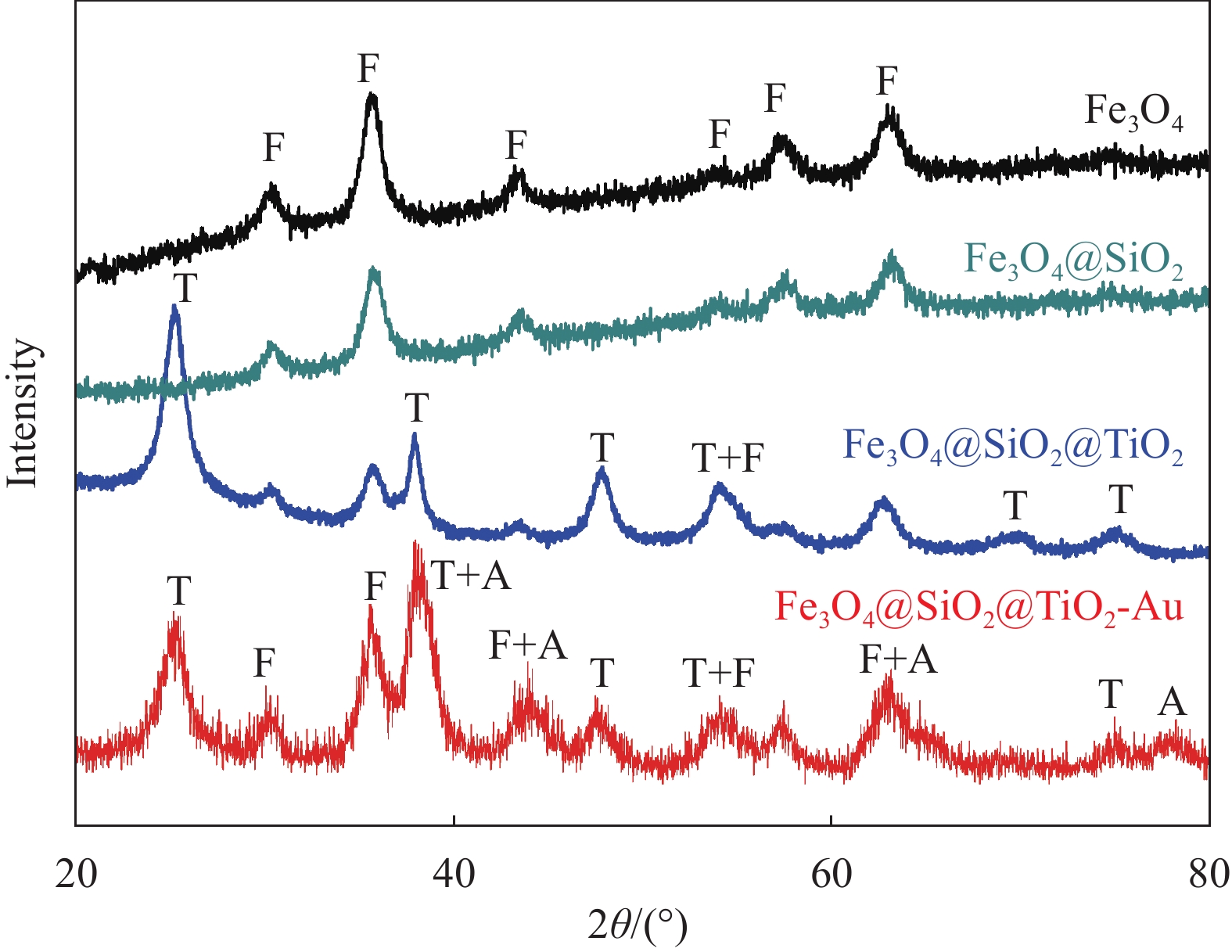
图4是Fe3O4、Fe3O4@SiO2、Fe3O4@SiO2@TiO2和Fe3O4@SiO2@TiO2-Au的XRD图谱。每个样品都出现了Fe3O4的衍射峰[16-17, 26],与面心立方相Fe3O4的标准卡片(JCPDS 19-0629) 匹配,在图中标记为F。包覆在Fe3O4表面的SiO2夹层并没有改变Fe3O4的结构,只是削弱了Fe3O4@SiO2的衍射峰强度,说明夹层SiO2是非晶态的。通过LPD法在Fe3O4@SiO2上沉积TiO2后,Fe3O4@SiO2@TiO2的图谱上出现了额外的衍射峰,其与锐钛矿型TiO2峰位一致(JCPDS Card No. 21-1272),在图中标记为 T 。Fe3O4@SiO2@TiO2-Au纳米复合物的图谱中出现了额外的低强度衍射峰,可以归入面心立方的Au (JCPDS 00-004-0784),在图中被标记为A。由于Au在复合材料中的含量低,且粒径小,对应于金的微弱衍射峰较弱。
![]() 图 4 Fe3O4、Fe3O4@SiO2、Fe3O4@SiO2@TiO2 和 Fe3O4@ SiO2@TiO2-Au的XRD图谱(F、T 和A分别表示Fe3O4、TiO2和Au的特征衍射峰)Figure 4. XRD patterns of Fe3O4, Fe3O4@SiO2, Fe3O4@SiO2 @TiO2 and Fe3O4@SiO2@TiO2-Au (F, T and A represent the diffraction peaks of Fe3O4, TiO2 and Au, respectively)
图 4 Fe3O4、Fe3O4@SiO2、Fe3O4@SiO2@TiO2 和 Fe3O4@ SiO2@TiO2-Au的XRD图谱(F、T 和A分别表示Fe3O4、TiO2和Au的特征衍射峰)Figure 4. XRD patterns of Fe3O4, Fe3O4@SiO2, Fe3O4@SiO2 @TiO2 and Fe3O4@SiO2@TiO2-Au (F, T and A represent the diffraction peaks of Fe3O4, TiO2 and Au, respectively)2.3 Fe3O4@SiO2@TiO2-Au的元素分析
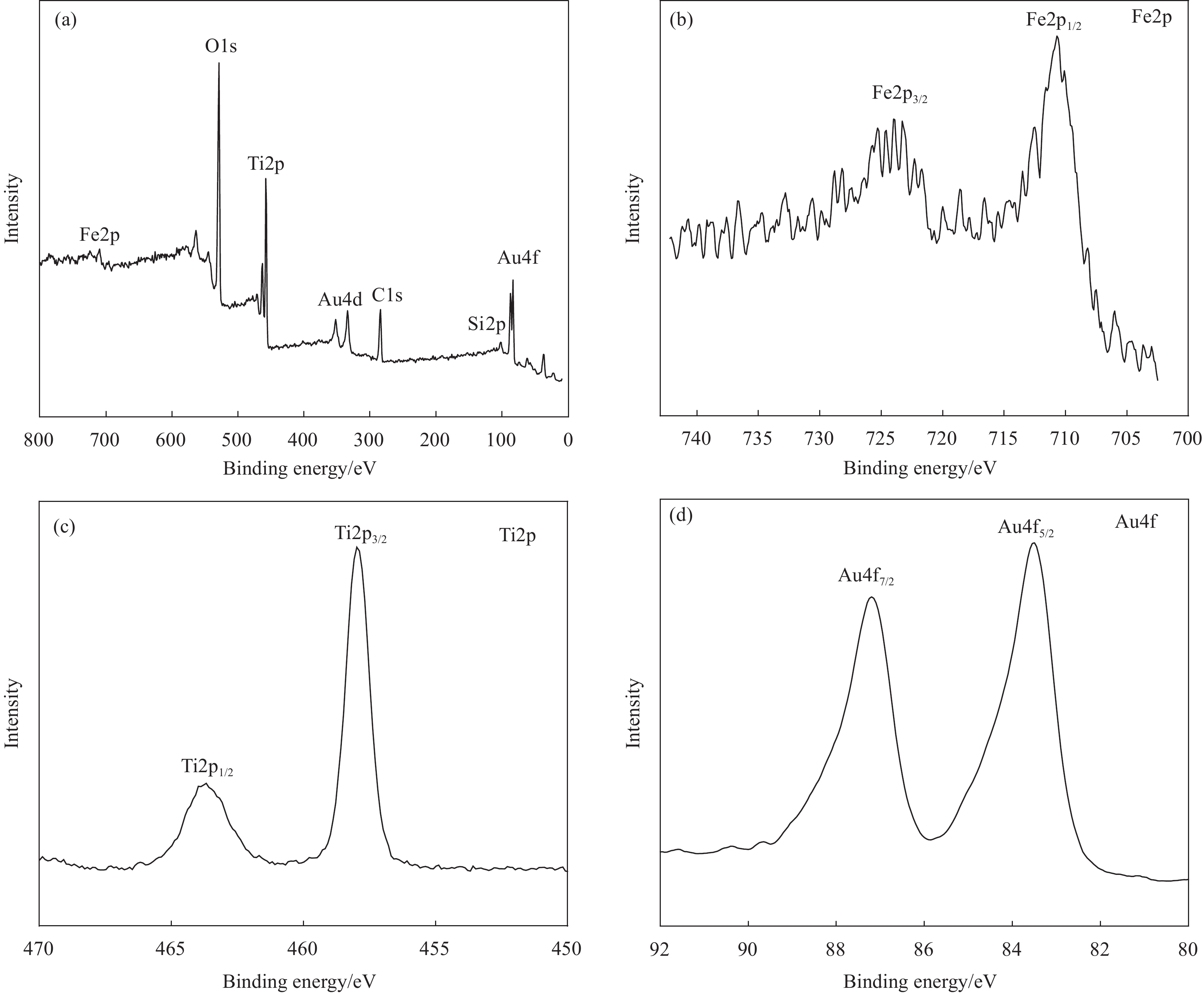
为了研究Fe3O4@SiO2@TiO2-Au的表面组成以及Fe、Ti和Au的价态,图5给出了Fe3O4@SiO2@TiO2-Au的XPS分析结果。从样品的XPS全谱中可以观察到Fe、O、Si、Ti和Au元素的特征峰(图5(a)),表明Fe3O4@SiO2@TiO2-Au是由这5种元素组成,这一结果与EDX分析一致。图5(b)~5(d)为Fe、Ti和Au的高分辨XPS图谱。图5(b)包含两个峰,分别对应于Fe2p3/2和Fe2p1/2,结合能为~710 eV和~724 eV。这些峰位归因于Fe3O4磁性核的存在[19-20, 26]。Ti2p的XPS图谱中464.2 eV和458.3 eV的峰(图5(c)),分别对应于Ti2p1/2和Ti2p3/2。这一结果表明了通过液相沉积形成的TiO2为锐钛矿型[27, 30]。图5(d)为复合微球中Au的高分辨XPS谱。位于83.8 eV和87.5 eV的峰分别对应Au04f7/2和Au04f5/2 [15, 19-20],表明Fe3O4@SiO2@TiO2表面沉积的Au NPs以金属单质的形式存在。
![]() 图 5 Fe3O4@SiO2@TiO2-Au的XPS全谱(a)和组成元素的高分辨XPS图谱(Fe2p (b)、Ti2p (c)和Au4f (d))Figure 5. XPS survey spectra of composite survey (a) and high-resolution spectra of Fe2p (b), Ti2p (c) and Au4f (d) of Fe3O4@SiO2@TiO2-Au
图 5 Fe3O4@SiO2@TiO2-Au的XPS全谱(a)和组成元素的高分辨XPS图谱(Fe2p (b)、Ti2p (c)和Au4f (d))Figure 5. XPS survey spectra of composite survey (a) and high-resolution spectra of Fe2p (b), Ti2p (c) and Au4f (d) of Fe3O4@SiO2@TiO2-Au2.4 Fe3O4@SiO2@TiO2-Au的磁性能
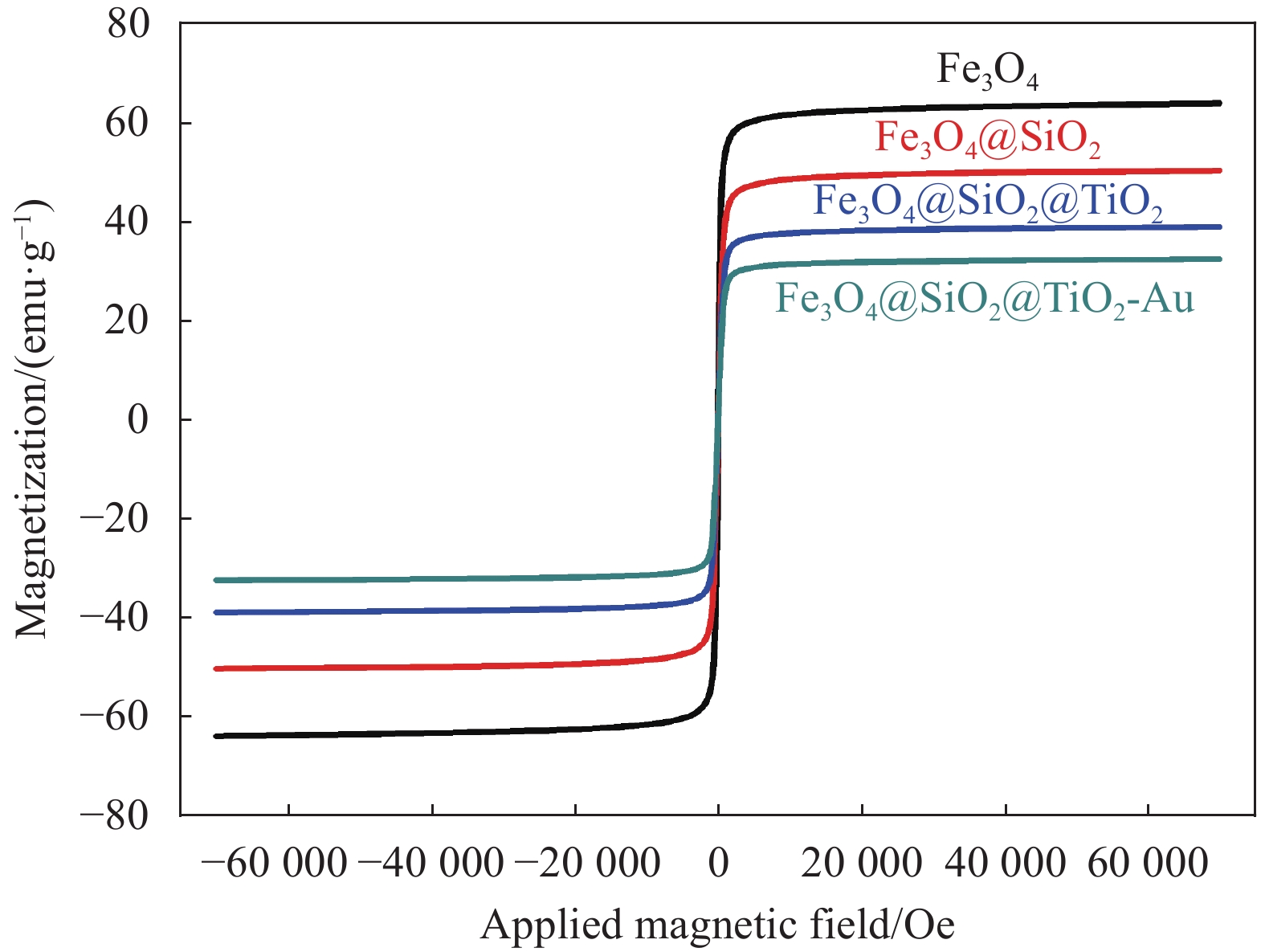
图6为Fe3O4、Fe3O4@SiO2、Fe3O4@SiO2@TiO2和Fe3O4@SiO2@TiO2-Au的室温磁滞回线,所有这4个样品都表现出超顺磁性能,没有明显的剩磁和矫顽力。其超顺磁性是由于Fe3O4内核中的小磁性纳米晶体产生的。Fe3O4的磁饱和强度(Ms)为63.5 emu·g−1。Fe3O4@SiO2和Fe3O4@SiO2@TiO2的Ms分别下降到50.5和39.3 emu·g−1,Fe3O4@SiO2@TiO2-Au的Ms进一步降低到29.6 emu·g−1。磁饱和强度的下降是由于在磁性Fe3O4上负载了非磁性涂层(TiO2、SiO2和Au NPs),减少了复合材料中的Fe3O4含量[19-21]。然而,Fe3O4@SiO2@TiO2-Au的磁性仍很高,足以用磁铁进行磁分离。
![]() 图 6 Fe3O4、Fe3O4@SiO2、Fe3O4@SiO2@TiO2和 Fe3O4@ SiO2@TiO2-Au的室温磁滞回线Figure 6. Room-temperature magnetization hysteresis loops of Fe3O4, Fe3O4@SiO2, Fe3O4@SiO2@TiO2 and Fe3O4@SiO2@TiO2-Au
图 6 Fe3O4、Fe3O4@SiO2、Fe3O4@SiO2@TiO2和 Fe3O4@ SiO2@TiO2-Au的室温磁滞回线Figure 6. Room-temperature magnetization hysteresis loops of Fe3O4, Fe3O4@SiO2, Fe3O4@SiO2@TiO2 and Fe3O4@SiO2@TiO2-Au2.5 Fe3O4@SiO2@TiO2-Au的光学性质
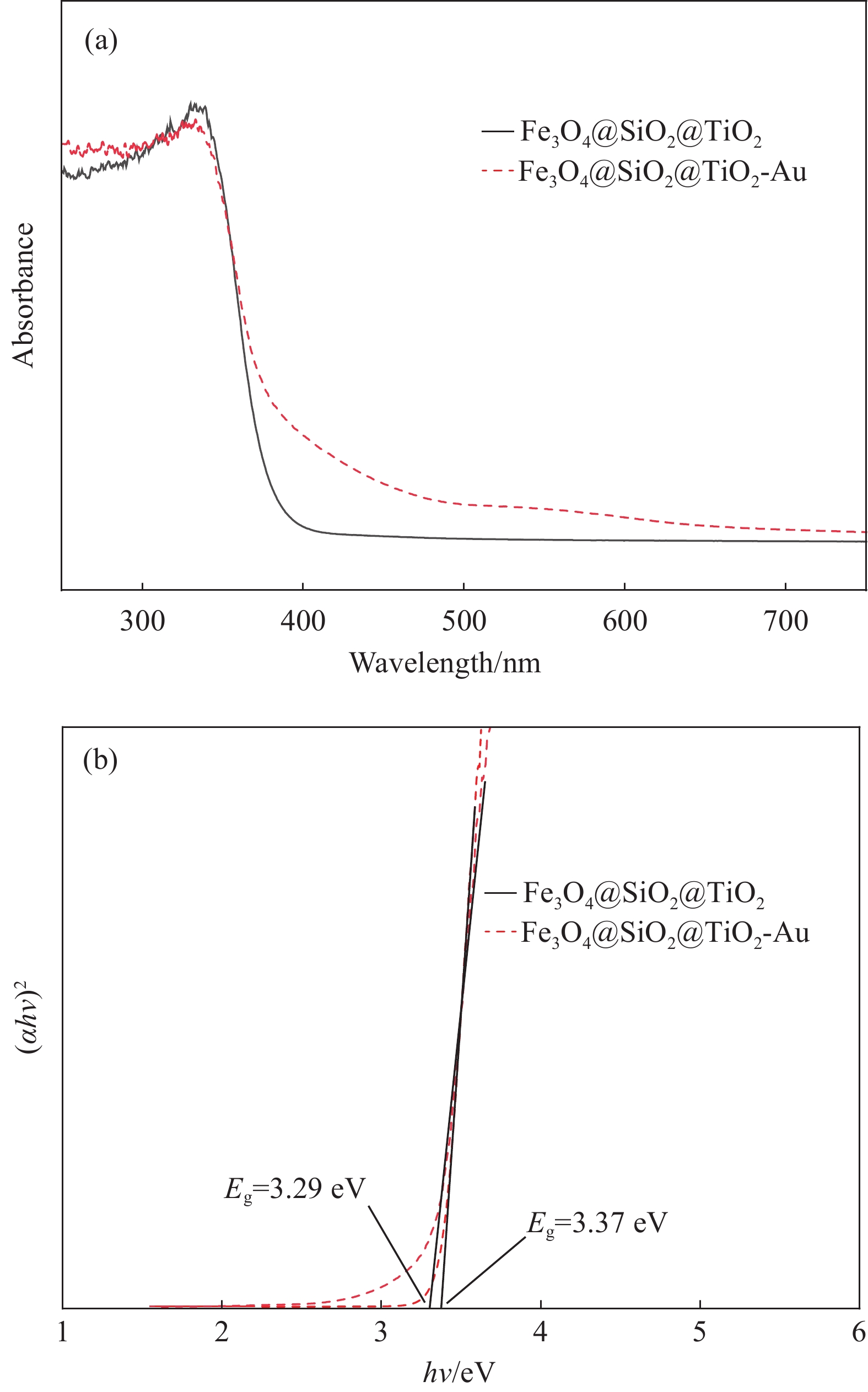
通过紫外-可见光漫反射可以研究样品的光吸收特性,如图7 所示。Fe3O4@SiO2@TiO2和Fe3O4@SiO2@TiO2-Au在 200~400 nm的紫外区显示出强烈的吸收(图7(a)),这是TiO2的特性吸收。Fe3O4@SiO2@TiO2在可见光区没有吸收,其吸收边缘约为 380 nm附近,根据Kubelka-Munk计算其带隙为3.37 eV (图7(b))。在TiO2上沉积金纳米粒子后,Fe3O4@SiO2@TiO2-Au的吸收边缘向可见光区发生了20 nm的微小红移。这种红移可能是由金与TiO2纳米粒子的相互作用引起的。同时与Fe3O4@SiO2@TiO2相比,Fe3O4@SiO2@TiO2-Au在可见光区域的吸光度增强了,其带隙也降低为3.29 eV (图7(b)),这都有助于增强其光催化能力。
![]() 图 7 Fe3O4@SiO2@TiO2和Fe3O4@SiO2@TiO2-Au紫外-可见漫反射光谱(a)和能带图(b)Figure 7. UV-Vis diffuse reflectance spectra (a) and band gap energies (b) of Fe3O4@SiO2@TiO2 and Fe3O4@SiO2@TiO2-Auα—Absorption coefficient; hν—Photon energy; Eg—Band gap energy
图 7 Fe3O4@SiO2@TiO2和Fe3O4@SiO2@TiO2-Au紫外-可见漫反射光谱(a)和能带图(b)Figure 7. UV-Vis diffuse reflectance spectra (a) and band gap energies (b) of Fe3O4@SiO2@TiO2 and Fe3O4@SiO2@TiO2-Auα—Absorption coefficient; hν—Photon energy; Eg—Band gap energy2.6 Fe3O4@SiO2@TiO2-Au的光催化性能及光催化机制
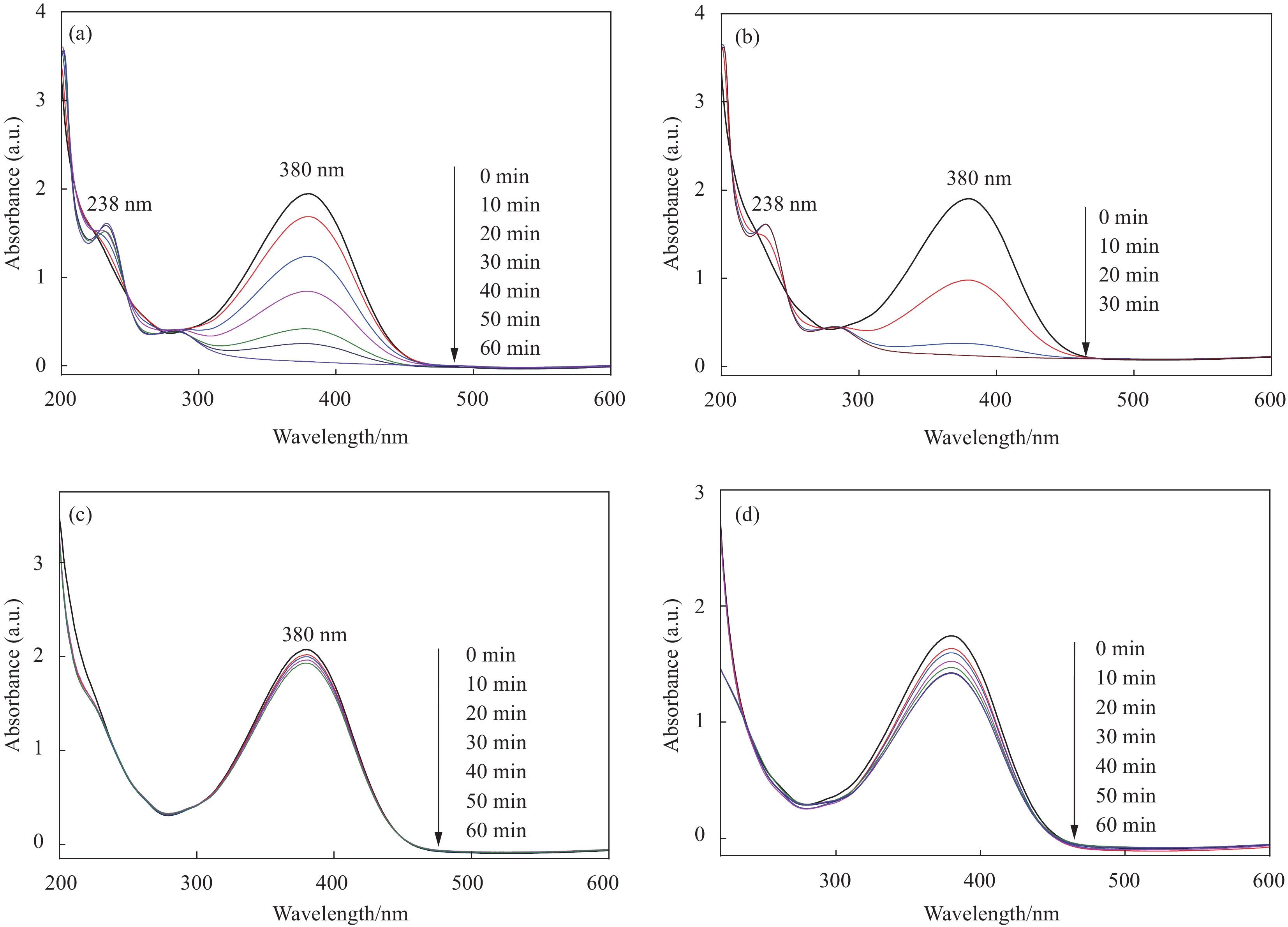
在HCOONH4存在下,Fe3O4@SiO2@TiO2和Fe3O4@SiO2@TiO2-Au光催化还原p-NA的UV-Vis吸收光谱如图8(a)和图8(b)所示。在这两种情况下,p-NA在380 nm处的特征吸收峰强度随着光照时间的延长而逐渐降低,直至完全消失。于此同时,在238 nm处出现了一个新的吸收峰,其强度逐渐增加直到达到最大值并保持不变。在238 nm处出现的峰是p-PDA的特征吸收峰[31-32],表明在光催化还原p-NA至p-PDA的过程中没有产生中间产物,如对亚硝基苯胺和对羟基氨基苯胺[32-34],p-NA能被这两个光催化剂定量地加氢还原成p-PDA,其转化率均为100%。然而在相同条件下,当Fe3O4@SiO2@TiO2-Au被用作光催化剂时(图8(b)),p-NA的吸收峰强度下降更为迅速,在30 min内几乎完全消失,而Fe3O4@SiO2@TiO2光催化剂将p-NA完全还原至p-PDA需要60 min,表明均匀、细小的Au NPs的引入能显著提高Fe3O4@SiO2@TiO2的光催化活性。通过6电子光催化还原p-NA至p-PDA的反应如下式所示[33-34]:
![]() 图 8 Fe3O4@SiO2@TiO2 ((a), (c))和 Fe3O4@SiO2@TiO2-Au ((b), (d))分别在有HCOONH4和无HCOONH4条件下光催化还原对硝基苯胺(p-NA)的UV-vis图谱(p-NA:20 mg·L−1;HCOONH4:800 mg·L−1;催化剂:50 mg)Figure 8. UV-vis absorption spectra of the p-nitroaniline (p-NA) aqueous solution under UV irradiation catalyzed by Fe3O4@SiO2@TiO2 ((a), (c)) in both the presence and the absence of HCOONH4 and Fe3O4@SiO2@TiO2-Au ((b), (d)) in both the presence and the absence of HCOONH4, respectively (p-NA: 20 mg·L−1; HCOONH4: 800 mg·L−1; Catalyst: 50 mg)
图 8 Fe3O4@SiO2@TiO2 ((a), (c))和 Fe3O4@SiO2@TiO2-Au ((b), (d))分别在有HCOONH4和无HCOONH4条件下光催化还原对硝基苯胺(p-NA)的UV-vis图谱(p-NA:20 mg·L−1;HCOONH4:800 mg·L−1;催化剂:50 mg)Figure 8. UV-vis absorption spectra of the p-nitroaniline (p-NA) aqueous solution under UV irradiation catalyzed by Fe3O4@SiO2@TiO2 ((a), (c)) in both the presence and the absence of HCOONH4 and Fe3O4@SiO2@TiO2-Au ((b), (d)) in both the presence and the absence of HCOONH4, respectively (p-NA: 20 mg·L−1; HCOONH4: 800 mg·L−1; Catalyst: 50 mg)H2NC6H4NO2+6e−+6H+→H2NC6H4NH2+2H2O (1) 对照实验(有光照而无光催化剂的p-NA+ HCOONH4和无光照的p-NA+HCOONH4+光催化剂)结果显示,p-NA的特征吸收峰强度几乎保持不变,既没有消耗p-NA,也没有产生p-PDA,表明即使加入HCOONH4,光催化剂和紫外光照射对于将p-NA还原成p-PDA是必不可少的,该反应是通过光催化而非热催化还原过程进行的。在无HCOONH4的情况下,分别研究了Fe3O4@SiO2@TiO2和Fe3O4@SiO2@TiO2-Au的光催化性能。图8(c)和图8(d)显示,在紫外光照射60 min后,p-NA向p-PDA的转化率很小,表明将p-NA通过光催化还原至p-PDA是由Fe3O4@SiO2@TiO2或Fe3O4@SiO2@TiO2-Au与HCOONH4协同催化的结果。HCOONH4作为空穴清除剂或牺牲性电子供体,对于提高光催化还原效率至关重要,由于它不仅作为空穴捕获剂可以迅速清除h+,有效地促进光催化剂中光生e−-h+对的分离,而且还可以提供大量的e−和H+,通过H+与光e−的还原作为H供体,促进硝基光催化氢化为胺基。可以看出,HCOONH4对光生h+的清除应是按照式(2)和式(3)进行[34-35]。
NH+4+8h++3H2O→NO−3+10H+ (2) HCO−2+2h+→CO2+H+ (3) 根据式(2)和式(3)的化学计量关系,通过光生h+氧化HCOONH4产生11个H+,几乎是p-NA 6电子还原式(1)消耗H+的两倍。在Fe3O4@SiO2@TiO2或Fe3O4@SiO2@TiO2-Au和HCOONH4存在下,测量了光催化反应前后溶液的pH值。在这两种反应条件下,反应介质的初始pH值约为5.6,而在反应完成后,pH值分别变为3.8和3.2,表明在HCOONH4氧化过程中,光生h+可以产生过量的H+。该结果表明紫外光照射和HCOONH4的存在下,Fe3O4@SiO2@TiO2光催化剂能诱导光生e−从TiO2转移到p-NA受体,而当Fe3O4@SiO2@TiO2有Au NPs表面修饰时,光生e−通过Au NPs从TiO2转移至p-NA变得更快,从而产生更多的H+,具有更高的光催化还原活性。
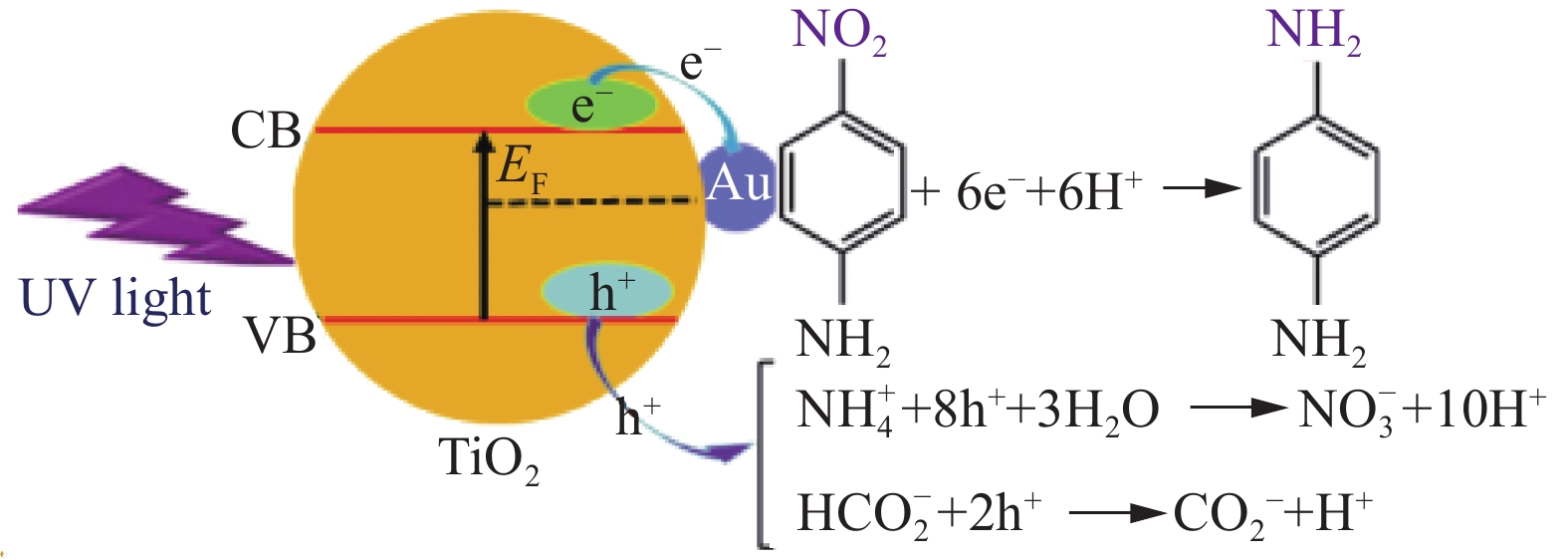
综上所述,对Fe3O4@SiO2@TiO2表面用分散均匀、粒径细小的Au NPs修饰可以显著提高其光催化还原p-NA的效率。图9为Fe3O4@SiO2@TiO2-Au光催化还原p-NA机制示意图,可以通过TiO2的能带结构和Au NPs的费米级解释Fe3O4@SiO2@TiO2-Au具有更高的光催化活性。Au NPs的费米能级(−5.1 eV)较TiO2费米能级(−4.3 eV)更低,因此在Au-TiO2异质结处可以形成一个肖特基势垒[15, 36-37]。在紫外光照射下,光生e−从TiO2价带(VB)被激发到导带(CB),在能量差驱动下可以克服肖特基势垒,然后自发地转移到Au的费米能级,有利于促进光生电荷载流子(h+和e−)的分离,从而提高了p-NA的光催化还原活性。此外,Fe3O4内核和TiO2外层之间的SiO2夹层可以避免Fe3O4和TiO2直接接触,避免光电子从TiO2转移到Fe3O4的低位CB,在界面上发生光溶解[19]。如果不引入Au NPs,来自TiO2的光激发的e−-h+对很容易发生复合,因此Fe3O4@SiO2@TiO2复合光催化剂的光催化活性较低。
![]() 图 9 Au NPs和TiO2之间的电荷转移和紫外光照下Fe3O4@SiO2@TiO2-Au催化还原p-NA的机制示意图Figure 9. Schematic diagram indicating the charge transfer between Au NPs and TiO2 and the mechanism for reduction of p-NA over the Fe3O4@SiO2@TiO2-Au photocatalyst under UV light irradiationEF—Fermi level of Au; CB—Conduction band; VB—Valence band
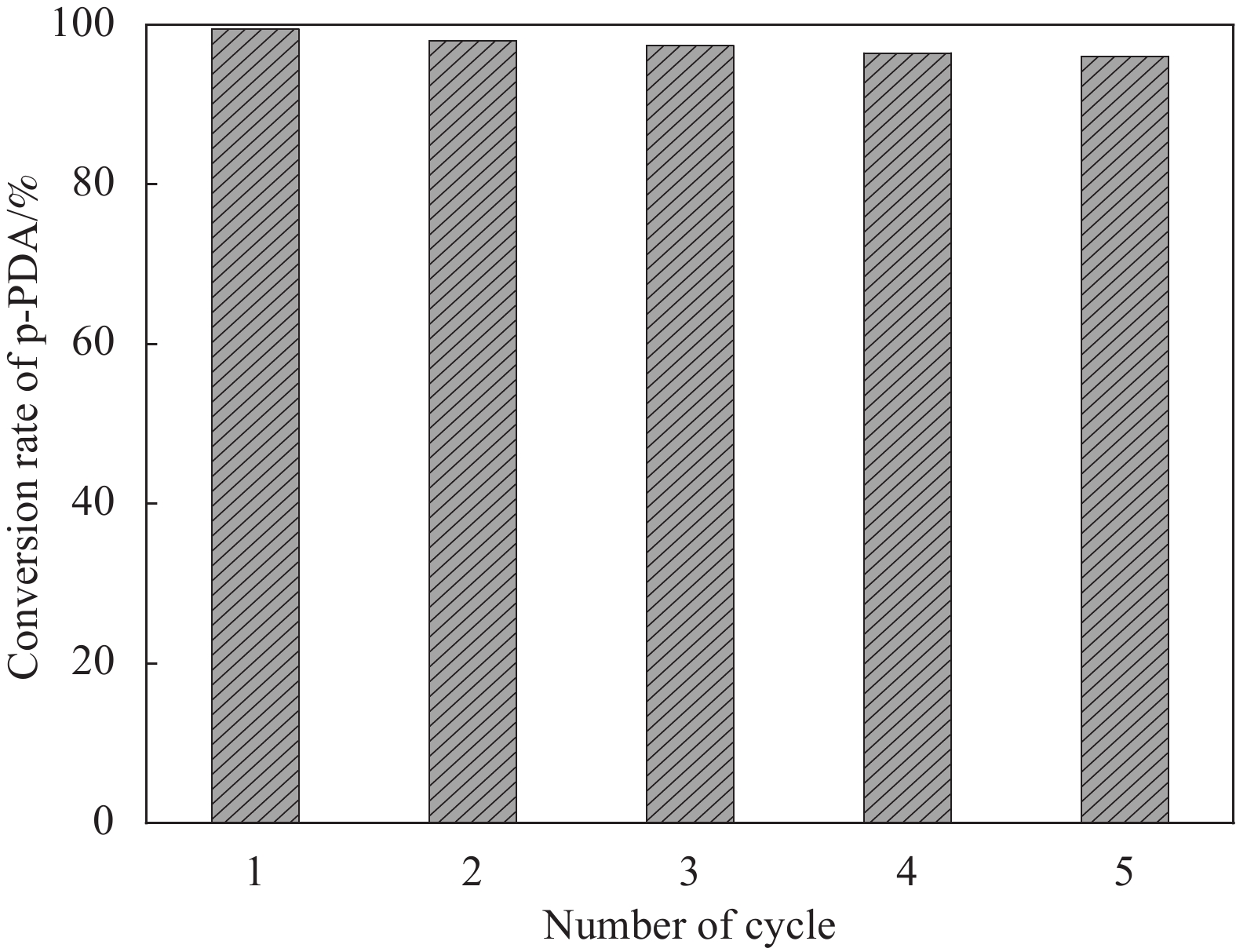
图 9 Au NPs和TiO2之间的电荷转移和紫外光照下Fe3O4@SiO2@TiO2-Au催化还原p-NA的机制示意图Figure 9. Schematic diagram indicating the charge transfer between Au NPs and TiO2 and the mechanism for reduction of p-NA over the Fe3O4@SiO2@TiO2-Au photocatalyst under UV light irradiationEF—Fermi level of Au; CB—Conduction band; VB—Valence band通过循环实验检测了催化剂的稳定性。第一次实验完成后,使用磁分离将Fe3O4@SiO2@TiO2-Au光催化剂从反应溶液中分离出来,并用去离子水彻底清洗,以便进一步使用。为了使p-NA完全转化为p-PDA,每个循环实验的辐照时间为50 min。如图10所示,该催化剂在运行5个循环后保持了较高的光催化效率(p-NA的转化率为 96%),显示了优异的循环稳定性。
![]() 图 10 用HCOONH4作为空穴捕获剂,Fe3O4@SiO2@TiO2-Au光催化剂的循环使用性能测试(每组循环光照时间为50 min)Figure 10. Reusability of Fe3O4@SiO2@TiO2-Au photocatalyst using HCOONH4 as hole scavenger (The duration of UV irradiation in each cycle is 50 min)p-PDA—p-phenylenediamine
图 10 用HCOONH4作为空穴捕获剂,Fe3O4@SiO2@TiO2-Au光催化剂的循环使用性能测试(每组循环光照时间为50 min)Figure 10. Reusability of Fe3O4@SiO2@TiO2-Au photocatalyst using HCOONH4 as hole scavenger (The duration of UV irradiation in each cycle is 50 min)p-PDA—p-phenylenediamine3. 结 论
(1)通过在溶胶-凝胶法在Fe3O4上包覆核SiO2,然后利用液相沉积法负载有序的TiO2外壳得到核壳结构的Fe3O4@SiO2@TiO2,最后用种子生长法在其表面负载均匀分散的金纳米粒子(Au NPs),制备了可磁分离的Fe3O4@SiO2@TiO2-Au复合光催化剂。
(2)光催化结果表明,在氮气气氛和HCOONH4空穴捕获剂存在下,两者作为光催化剂都能将对硝基苯胺(p-NA)还原成对苯二胺(p-PDA)。然而与Fe3O4@SiO2@TiO2相比,Fe3O4@SiO2@TiO2-Au将p-NA完全还原到p-PDA需要更短的光照射时间,具有更优异的光催化活性。增强的光催化活性主要归因于TiO2上分散的Au NPs,可以作为电子捕获剂,能有效促进光电子从TiO2转移到被还原物p-NA,抑制了h+-e−对的重新结合,从而提高了光催化还原活性。
(3) TiO2-Au NPs优异的光催化活性和Fe3O4的超顺磁性相结合,不仅具有光催化还原其他含有可还原性基团的有机污染物的能力,而且在光催化反应后容易被磁分离和循环使用的优点。该研究为TiO2基光催化剂将具有不同官能团的硝基芳烃化合物光还原为氨基芳烃化合物提供了一个新的途径。
-
![]()
图 1 磁性Fe3O4@SiO2@TiO2-Au核壳结构微球的组装流程图
Figure 1. Schematic diagram for fabricating magnetic Fe3O4@SiO2@TiO2-Au microspheres with core-shell structure
NPs—Nanoparticles; EG—Ethylene glycol; TEOS—Tetraethyl orthosilicate
![]()
图 2 Fe3O4 ((a), (b))、Fe3O4@SiO2 ((c), (d))、Fe3O4@SiO2@TiO2 ((e), (f))、Au-seed-decorated Fe3O4@SiO2@TiO2 (g) 和 Fe3O4@SiO2@TiO2-Au ((h), (i))的不同放大倍数的TEM图像;(j) 沉积在Fe3O4@SiO2上的TiO2和Au纳米粒子(NPs)的高分辨透射电镜图像
Figure 2. TEM images with different magnifications of Fe3O4 ((a), (b)), Fe3O4@SiO2 ((c), (d)), Fe3O4@SiO2@TiO2 ((e), (f)), Au-seed-decorated Fe3O4@SiO2@TiO2 (g), Fe3O4@SiO2@TiO2-Au ((h), (i)); (j) HRTEM of the deposited TiO2 and Au nanoparticles (NPs) on Fe3O4@SiO2
![]()
图 3 Fe3O4@SiO2@TiO2-Au复合物的STEM图像(a)、元素谱分布图((b)~(f))、样品的能量色散谱(g)
Figure 3. STEM image (a), a series of EDS elemental mapping images (Fe, Si, Ti, O and Au) ((b)-(f)), its corresponding EDX spectrum (g) of Fe3O4@SiO2@TiO2-Au composite
![]()
图 4 Fe3O4、Fe3O4@SiO2、Fe3O4@SiO2@TiO2 和 Fe3O4@ SiO2@TiO2-Au的XRD图谱(F、T 和A分别表示Fe3O4、TiO2和Au的特征衍射峰)
Figure 4. XRD patterns of Fe3O4, Fe3O4@SiO2, Fe3O4@SiO2 @TiO2 and Fe3O4@SiO2@TiO2-Au (F, T and A represent the diffraction peaks of Fe3O4, TiO2 and Au, respectively)
![]()
图 5 Fe3O4@SiO2@TiO2-Au的XPS全谱(a)和组成元素的高分辨XPS图谱(Fe2p (b)、Ti2p (c)和Au4f (d))
Figure 5. XPS survey spectra of composite survey (a) and high-resolution spectra of Fe2p (b), Ti2p (c) and Au4f (d) of Fe3O4@SiO2@TiO2-Au
![]()
图 6 Fe3O4、Fe3O4@SiO2、Fe3O4@SiO2@TiO2和 Fe3O4@ SiO2@TiO2-Au的室温磁滞回线
Figure 6. Room-temperature magnetization hysteresis loops of Fe3O4, Fe3O4@SiO2, Fe3O4@SiO2@TiO2 and Fe3O4@SiO2@TiO2-Au
![]()
图 7 Fe3O4@SiO2@TiO2和Fe3O4@SiO2@TiO2-Au紫外-可见漫反射光谱(a)和能带图(b)
Figure 7. UV-Vis diffuse reflectance spectra (a) and band gap energies (b) of Fe3O4@SiO2@TiO2 and Fe3O4@SiO2@TiO2-Au
α—Absorption coefficient; hν—Photon energy; Eg—Band gap energy
![]()
图 8 Fe3O4@SiO2@TiO2 ((a), (c))和 Fe3O4@SiO2@TiO2-Au ((b), (d))分别在有HCOONH4和无HCOONH4条件下光催化还原对硝基苯胺(p-NA)的UV-vis图谱(p-NA:20 mg·L−1;HCOONH4:800 mg·L−1;催化剂:50 mg)
Figure 8. UV-vis absorption spectra of the p-nitroaniline (p-NA) aqueous solution under UV irradiation catalyzed by Fe3O4@SiO2@TiO2 ((a), (c)) in both the presence and the absence of HCOONH4 and Fe3O4@SiO2@TiO2-Au ((b), (d)) in both the presence and the absence of HCOONH4, respectively (p-NA: 20 mg·L−1; HCOONH4: 800 mg·L−1; Catalyst: 50 mg)
![]()
图 9 Au NPs和TiO2之间的电荷转移和紫外光照下Fe3O4@SiO2@TiO2-Au催化还原p-NA的机制示意图
Figure 9. Schematic diagram indicating the charge transfer between Au NPs and TiO2 and the mechanism for reduction of p-NA over the Fe3O4@SiO2@TiO2-Au photocatalyst under UV light irradiation
EF—Fermi level of Au; CB—Conduction band; VB—Valence band
-
[1] AHMED A, SANDLER S I. Hydration free energies of multifunctional nitroaromatic compounds[J]. Journal of Chemical Theory and Compution, 2013, 9(6): 2774-2785. DOI: 10.1021/ct3011002
[2] ALINA G, SÜHNHOLZ S, GEORGI A, et al. Fe-zeolites for the adsorption and oxidative degradation of nitroaromatic compounds in water[J]. Journal of Hazardous Materials, 2023, 459: 132125. DOI: 10.1016/j.jhazmat.2023.132125
[3] BILAL M, BAGHERI A R, BHATT P, et al. Environmental occurrence, toxicity concerns, and remediation of recalcitrant nitroaromatic compounds[J]. Journal of Environmental Management, 2021, 291: 112685 DOI: 10.1016/j.jenvman.2021.112685
[4] HIDALGO A M, LEÓN G, GÓMEEZ M, et al. Behaviour of RO90 membrane on the removal of 4-nitrophenol and 4-nitroaniline by low pressure reverse osmosis[J]. Journal of Water Process Engineering, 2015, 7: 169-175. DOI: 10.1016/j.jwpe.2015.06.007
[5] ZHU C C, HUANG H N, CHEN Y G. Recent advances in biological removal of nitroaromatics from wastewater[J]. Environmental Pollution, 2022, 307: 119570.
[6] 殷月月, 杨勇, 张良柱, 等. 金/钯哑铃状纳米晶的制备及其催化对硝基苯酚还原研究[J]. 无机材料学报, 2018, 33(1): 19-26. DOI: 10.15541/jim20170146 YIN Yueyue, YANG Yong, ZHANG Liangzhu, et al. Facile synthesis of Au/Pd nano-dumbells for catalytic reduction of p-nitrophenol[J]. Journal of Inorganic Materials, 2018, 33(1): 19-26(in chinese). DOI: 10.15541/jim20170146
[7] YANG M Q, PAN X Y, ZHANG N, et al. A facile one-step way to anchor noble metal (Au, Ag, Pd) nanoparticles on a reduced graphene oxide mat with catalytic activity for selective reduction of nitroaromatic compounds[J]. CrystEngComm, 2013, 15(34): 6819-6828. DOI: 10.1039/c3ce40694f
[8] LUO P F, XU K L, ZHANG R, et al. Highly efficient and selective reduction of nitroarenes with hydrazine over supported rhodium nanoparticles[J]. Catalysis Science & Technology, 2012, 2(2): 301-304.
[9] FOUNTOULAKI S, DAIKOPOULOU V, GKIZIS P L, et al. Mechanistic studies of the reduction of nitroarenes by NaBH4 or hydrosilanes catalyzed by supported gold nanoparticles[J]. ACS Catalysis, 2014, 4(10): 3504-3511. DOI: 10.1021/cs500379u
[10] GONG Y Z, YANG C, JI H W, et al. Aqueous oxidations started by TiO2 photoinduced holes can be a rate-determining step[J]. Chemistry—An Asian Journal, 2017, 12(16): 2048-2051. DOI: 10.1002/asia.201700658
[11] PAN D L, HAN Z Y, MIAO Y C, et al. Thermally stable TiO2 quantum dots embedded in SiO2 foams: Characterization and photocatalytic H2 evolution activity[J]. Applied Catalysis B: Environmental, 2018, 229: 130-138. DOI: 10.1016/j.apcatb.2018.02.022
[12] YANG Y, WANG G Z, DENG Q, et al. Microwave-assisted fabrication of nanoparticulate TiO2 microspheres for synergistic photocatalytic removal of Cr(VI) and methyl orange[J]. ACS Applied Materials & Interfaces, 2014, 6(4): 3008-3015.
[13] LI W, WU Z X, WANG J X, et al. A perspective on mesoporous TiO2 materials[J]. Chemistry of Materials, 2014, 26(1): 287-298. DOI: 10.1021/cm4014859
[14] LIU Q, QIU W, WU P, et al. Low-temperature ammonia oxidation in a microchannel reactor with wall-loaded X (X = Pt, Pd, Rh, PtPdRh)/TiO2 nanotube catalysts[J]. Industrial & Engineering Chemistry Research, 2019, 58(23): 9819-9828.
[15] HUSSAIN M, AHMAD M, NISAR A, et al. Enhanced photocatalytic and electrochemical properties of Au nanoparticles supported TiO2 microspheres[J]. New Journal of Chemistry, 2014, 38(4): 1424-1432.
[16] 龚新怀, 李明春, 杨坤, 等. 纳米Fe3O4@茶渣/海藻酸钙磁性复合材料制备及其对亚甲基蓝的吸附性能与吸附机制[J]. 复合材料学报, 2021, 38(2): 424-438. GONG Xinhuai, LI Mingchun, YANG Kun, et al. Preparation of nano-Fe3O4@tea waste/calcium alginate magnetic composited bead and it's adsorption characteristics and mechanisms for methylene blue from aqueous solution[J]. Acta Materiae Compositae Sinica, 2021, 38(2): 424-438(in Chinese).
[17] 梁艳莉, 马剑琪, 郭少波. CoFe2O4@PDA@Pt核壳型磁性复合材料的制备及催化性能[J]. 复合材料学报, 2021, 38(5): 1551-1557. LIANG Yanli, MA Jianqi, GUO Shaobo. Preparation and catalytic properties of CoFe2O4@PDA@Pt magnetic composite with core shell structure[J]. Acta Materiae Compositae Sinica, 2021, 38(5): 1551-1557(in Chinese).
[18] 方丽, 罗琨. Fe3O4@Ag3PO4光催化剂可见光降解盐酸环丙沙星的研究[J]. 水处理技术, 2022, 48(12): 71-76. FANG Li, LUO Kun. Photocatalytic degradation of ciprofloxacin hydrochloride by Fe3O4@Ag3PO4 under visible-light irradiation[J]. Technology of Water Treatment, 2022, 48(12): 71-76(in Chinese).
[19] JIN S, PARK E, GUO S, et al. Process monitoring of photocatalytic degradation of 2, 4-dinitrotoluene by Au-decorated Fe3O4@TiO2 nanoparticles: Surface-enhanced Raman scattering method[J]. Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy, 2022, 275: 121155. DOI: 10.1016/j.saa.2022.121155
[20] LI C Y, YOUNESI R, CAI Y L, et al. Photocatalytic and antibacterial properties of Au-decorated Fe3O4@mTiO2 core-shell microspheres[J]. Applied Catalysis B: Environmental, 2014, 156-157: 314-322. DOI: 10.1016/j.apcatb.2014.03.031
[21] CHI Y, YUAN Q, LI Y J, et al. Magnetically separable Fe3O4@SiO2@TiO2-Ag microspheres with well-designed nanostructure and enhanced photocatalytic activity[J]. Journal of Hazardous Materials, 2013, 262: 404-411. DOI: 10.1016/j.jhazmat.2013.08.077
[22] LI X Y, LIU D P, SONG S Y, et al. Fe3O4@SiO2@TiO2@Pt hierarchical core-shell microspheres: Controlled synthesis, enhanced degradation system, and rapid magnetic separation to recycle[J]. Crystal Growth & Design, 2014, 14(11): 5506-5511.
[23] MA D W, SCHNEIDER J, LEE W I, et al. Controllable synthesis and self-template phase transition of hydrous TiO2 colloidal spheres for photo/electrochemical applications[J]. Advances in Colloid and Interface Science, 2021, 295: 102493. DOI: 10.1016/j.cis.2021.102493
[24] BEYDOUM D, AMAL R, LOW G, et al. Occurrence and prevention of photo dissolution at the phase junction of magnetite and titanium dioxide[J]. Journal of Molecular Catalysis A: Chemical, 2002, 180(1-2): 193-200. DOI: 10.1016/S1381-1169(01)00429-0
[25] KALIDINDI S B, JAGIRDAR B R. Nanocatalysis and prospects of green chemistry[J]. ChemSusChem, 2012, 5(1): 65-75. DOI: 10.1002/cssc.201100377
[26] CHENG C M, WEN Y H, XU X F, et al. Tunable synthesis of carboxyl-functionalized magnetite nanocrystal clusters with uniform size[J]. Journal of Materials Chemistry, 2009, 19(46): 8782-8788. DOI: 10.1039/b910832g
[27] MA J Q, GUO S B, GUO X H, et al. Liquid-phase deposition of TiO2 nanoparticles on core-shell Fe3O4@SiO2 spheres: Preparation, characterization, and photocatalytic activity[J]. Journal of Nanoparticle Research, 2015, 17(7): 307. DOI: 10.1007/s11051-015-3107-1
[28] JANA N R, GEARHEART L, MURPHY C J. Seeding growth for size control of 5-40 nm diameter gold nanoparticles[J]. Langmuir, 2001, 17(22): 6782-6786. DOI: 10.1021/la0104323
[29] ZHANG Y Y, GUO S B, MA J Q, et al. Preparation, characterization, catalytic performance and antibacterial activity of Ag photodeposited on monodisperse ZnO submicron spheres[J]. Journal of Sol-Gel Science and Technology, 2014, 72(1): 171-178. DOI: 10.1007/s10971-014-3440-3
[30] LI G, BAI R B, ZHAO X S. Coating of TiO2 thin films on the surface of SiO2 microspheres: Toward industrial photocatalysis[J]. Industrial & Engineering Chemistry Research, 2008, 47(21): 8228-8232.
[31] CHIU C Y, CHUNG P J, LAO K U, et al. Facet-dependent catalytic activity of gold nanocubes, octahedra, and rhombic dodecahedra toward 4-nitroaniline reduction[J]. Journal of Physical Chemistry C, 2012, 116(44): 23757-23763. DOI: 10.1021/jp307768h
[32] WU W, WEN L, SHEN L, et al. A new insight into the photocatalytic reduction of 4-nitroaniline to p-phenylenediamine in the presence of alcohols[J]. Applied Catalysis B: Environmental, 2013, 130-131: 163-167.
[33] YAMAMOTO Y, FUKUI M, TANAKA A, et al. Hydrogen- and noble metal-free conversion of nitro aromatics to amino aromatics having reducible groups over an organically modified TiO2 photocatalyst under visible light irradiation[J]. Catalysis Science & Technology, 2019, 9(4): 966-973.
[34] FUKUI M, KOSHIDA W, TANAKA A, et al. Photocatalytic hydrogenation of nitrobenzenes to anilines over noble metal-free TiO2 utilizing methylamine as a hydrogen donor[J]. Applied Catalysis B: Environmental, 2020, 268: 118446. DOI: 10.1016/j.apcatb.2019.118446
[35] CHENTHAMARKSHAN C R, RAJESHWAR K, WOLFRUM E R. Heterogeneous photocatalytic reduction of Cr(VI) in UV-irradiated titania suspensions: Effect of protons, ammonium ions, and other interfacial aspects[J]. Langmuir, 2000, 16(6): 2715-2721. DOI: 10.1021/la9911483
[36] JAKOB M, LEVANON H, KAMAT P V. Charge distribution between UV-irradiated TiO2 and gold nanoparticles: Determination of shift in the Fermi level[J]. Nano Letters, 2003, 3(3): 353-358. DOI: 10.1021/nl0340071
[37] VANDARKUZHALI S A A, PUGAZHENTHIRAN N, MANGALARAJA R V, et al. Ultrasmall plasmonic nanoparticles decorated hierarchical mesoporous TiO2 as an efficient photocatalyst for photocatalytic degradation of textile dyes[J]. ACS Omega, 2018, 3(3): 9834-9845.
-
期刊类型引用(1)
1. 李佥,王添誉,孙西同,陈晓艺,李苗,韩雨擎,曾祥冰,孙芳鸿,李宪臻. 贻贝仿生修饰多孔磁性材料的制备及其在固定化脂肪酶中的应用. 复合材料学报. 2024(11): 6156-6169 .  本站查看
本站查看
其他类型引用(0)
-
目的
TiO基的光催化剂已被广泛用于光氧化降解各种有机物污染物和光还原水中六价铬Cr(VI)至三价铬Cr(Ⅲ)。然而,对光催化还原硝基芳香化合物为胺基芳香化合物的研究鲜有报道。本文制备FeO@SiO@TiO-Au磁性纳米复合光催化剂,以HCOONH作为空穴捕获剂和氢源,用光催化还原对硝基苯胺(p-NA)作模型反应,研究其催化性能。
方法首先采用溶剂热法制备磁性FeO亚微米球,然后通过sol-gel过程,在FeO亚微米球表面形成均匀光滑的SiO壳层,采用液相沉积(LPD)法将锐钛矿型TiO沉积在非晶SiO包覆的FeO上,制备了核壳结构的FeO@SiO@TiO磁性光催化剂。通过种子生长法将均匀分散的Au纳米粒子(Au NPs)修饰在其表面,以获得FeO@SiO@TiO-Au纳米复合材料。通过TEM、XRD和XPS等手段对催化剂结构进行了表征,并通过紫外-可见光漫反射谱研究了其带隙。在紫外光照射下,用HCOONH作为空穴捕集剂和H源,以对硝基苯胺(p-NA)光催化还原至对苯二胺(p-PDA)作为模型反应,评价催化剂的光催化还原性能。
结果结构表征结果可以看出,FeO呈现出球形结构,单分散性好,在其表面包覆了均匀光滑的SiO壳层 (厚度约30 nm);通过LPD法在FeO@SiO表面沉积了一层致密均匀的TiO NPs外壳,单分散的Au (平均粒径5.2 nm)牢固地负载在TiO的表面,形成核壳结构的FeO@SiO@TiO-Au复合球。STEM、XRD、XPS等表征也证明了这一结构的成功合成。样品的紫外-可见光漫反射光谱表明在TiO上沉积金纳米粒子后,FeO@SiO@TiO-Au的吸收边缘向可见光区发生了20 nm的红移。光催化结果表明,FeO@SiO@TiO和FeO@SiO@TiO-Au都能光催化还原p-NA至p-PDA,其转化率达到100%,FeO@SiO@TiO-Au消耗更少时间,催化效率更高。对照试验还表明,HCOONH作为空穴清除剂或牺牲性电子供体,对于提高光催化还原效率至关重要。
结论制备了可磁分离的FeO@SiO@TiO-Au纳米复合光催化剂。FeO@SiO@TiO-Au将p-NA完全还原到p-PDA需要更短的光照射时间,具有更优异的光催化活性。增强的光催化活性主要归因于TiO上分散的Au NPs,可以作为电子捕获剂,抑制了h-e对的重新结合,能有效促进光电子从TiO通过Au转移到被还原物p-NA,从而提高了光催化还原活性。
 下载:
下载:

计量
- 文章访问数: 474
- HTML全文浏览量: 366
- PDF下载量: 41
- 被引次数: 1
