

Preparation of CeO2/CdxZn1−xS photocatalyst and its high-performance photocatalytic hydrogen production
-
摘要:
采用溶剂热法制备了CdxZn1−xS (CZS-X)固溶体、CeO2/CdxZn1−xS (y%CCZS-X)异质结,并采用XRD、SEM、XPS等表征手段对其样品的晶型、形貌、结构、元素组成等进行了表征。可见光照射下,研究了CZS-X固溶体、y%CCZS-0.3异质结产氢性能。CZS-0.3异质结的产氢速率为3.86 mmol·g−1·h−1,分别是CdS、ZnS的4.85、11.03倍。10%CCZS-0.3异质结具有最佳的光催化性能,产氢速率为7.89 mmol·g−1·h−1,分别是CeO2、CZS-0.3固溶体的40.25、2.04倍。光照下,CeO2的电子迁移到CZS-X,使得靠近CeO2的异质结界面部分带正电,而靠近CZS-X的异质结界面部分带负电,形成内电场,增强了载流子分离与迁移性能。
-
关键词:
- CeO2/CdxZn1−xS /
- 固溶体 /
- 异质结 /
- 内电场 /
- 光催化产氢
Abstract:CdxZn1−xS (CZS-X) solid solution and CeO2/CdxZn1−xS (y%CCZS-X) heterojunction were prepared by solvothermal method, and the crystal shape, morphology, structure and elemental composition were characterized by XRD, SEM and XPS. The hydrogen production properties of CZS-X solid solution and y%CCZS-X heterojunction were studied under visible light irradiation. The hydrogen production rate of CZS-0.3 heterojunction was 3.86 mmol·g−1·h−1, which was 4.85 and 11.03 times that of CdS and ZnS, respectively. 10%CCZS-0.3 heterojunction had the best photocatalytic performance, and the hydrogen production rate was 7.89 mmol·g−1·h−1, which was 40.25 and 2.04 times of that of CeO2 and CZS-0.3 solid solutions, respectively. Under light, the electrons of CeO2 migrate to CZS-X, making the part of the heterojunction interface near CeO2 positively charged, while the part of the heterojunction interface near CZS-X negatively charged, forming an internal electric field, which enhanced the carrier separation and migration performance.
-
燃料电池是一种新型的电化学发电设备,被认为是解决化石燃料供应减少、环境污染和全球变暖等问题的潜在解决方案[1-2]。氧还原反应(ORR)是燃料电池不可或缺的阴极过程,但其本身是一个缓慢的动力学过程,严重阻碍了电池的大规模应用[3-4]。大量研究表明,商业铂碳是最高效的氧还原催化剂,能快速提升氧还原反应的效率,但地球上铂的资源稀少,价格昂贵,大量的商业化使用铂会导致电池成本高昂。因此,减少铂负载量或者制备高活性非铂催化剂具有非常重要的实际意义[5]。目前,从降低铂的使用量来说,人们开发了大量铂基ORR电催化剂,主要以铂和其它过渡金属如钴、镍、锰、铁、钯、银组成的铂基双金属或铂基多金属复合物纳米颗粒,其载体主要是碳材料[6-8];从完全弃用铂来说,目前开发的这种非铂ORR电催化剂种类繁多,但主要是以不同类型碳材料负载的镍、钴、铁、锰形成的复合物[9-11]。研究表明,这些新型的ORR电催化剂在碱性介质中一般表现出良好的电活性,在电池的实际应用中也表现稳定,但对于酸性环境的质子交换膜燃料电池而言,使用的介质是固体聚合物,通过质子的传递实现电荷的转移。对于非铂ORR电催化剂而言,目前还没有找到它们在酸性介质中的ORR电活性能够达到或接近金属铂的水平;此外,在实际应用中发现,质子交换膜燃料电池使用非铂类材料为阴极电催化剂时,在电池的运行过程中,由于电极处于静止状态,阴极在催化ORR时,产生的少量过氧化氢或过氧化离子不断在阴极表面积累,对非铂类材料的活性产生毒化效应,使催化剂活性产生较为明显的下降[8]。另一方面,如果在质子交换膜燃料电池中使用铂或铂基ORR电催化剂,虽然可以达到电池稳定运行的效果,但铂的实际使用量并没有明显降低,同样存在成本高昂的问题。
除此之外,对于市面上可用的Pt/C催化剂,其稳定性仍然是一个需要进一步解决的主要问题。杂原子可以有效地改变sp2共轭碳基基质上的电荷和自旋分布,促进氧的吸附,提高电催化效率[12-14]。可利用杂原子掺杂碳材料,如氮掺杂碳纳米管,或采用金属氧化物增强Pt粒子的稳定性[15]。此外,热解过渡金属含氮碳配合物(M-Nx/C)因其具有较高的活性而被认为是最有发展前景的化合物[16-17]。过渡金属和氮掺杂剂之间的配位可以有效的调控局部电子结构,提高导电性。铁、钴是资源丰富的非贵金属,作为双金属合金催化剂用于催化系统中具有独特的性能。
因此,制备低铂载量、ORR电活性优异且稳定的电催化剂,对于燃料电池的大规模实际应用具有重要意义。在本工作中,利用双氰胺作为碳源和氮源,二茂铁和酞菁钴分别作为铁源和钴源,通过简单的高温热解得到一系列不同铁钴原子负载比例的碳纳米片(Fe-Co-N/C),使其本身即具有良好的ORR活性;在此基础上进一步将少量Pt负载在Fe1-Co1-N/C上,并再次通过简单的热处理,得到低铂载量的、氮掺杂碳纳米片负载的铂-铁-钴复合材料(Fe1-Co1-Pt-N/C)。
1. 实验材料及方法
1.1 原材料
双氰胺、二茂铁、酞菁钴、四氯化铂购自阿拉丁化学试剂官网;商用Pt/C (40%,Johnson Matthey Corp.)购自上海群益能源设备有限公司;采用超纯水发生器生产实验用水(18.2 MΩ·cm);无水乙醇购自中国汕头西龙化工厂;Nafion溶液(5%,杜邦)购自国药控股化学试剂有限公司(上海)。
1.2 Fe-Co-N/C的制备
将100 mg二茂铁和300 mg酞菁钴混合(以摩尔比Fe∶Co=1∶1 为例),加入20 mL乙醇,搅拌,然后超声30 min,再加入3 g双氰胺,转移到研钵中,将混合物充分研磨均匀,之后放到烘箱在60℃下烘干,接着将混合物研磨成粉末状,转移到管式炉中,以4℃/min的升温速率,加热到600℃,保持2小时,再以同样的升温速率加热到800℃,继续保持2小时。最后得到负载铁钴金属颗粒的碳氮纳米片(Fe1-Co1-N/C).
催化剂Fe-Co-N/C中铁和钴摩尔比分别为1∶1、1∶2、2∶1和3∶1时,对应的二茂铁和酞菁钴用量分别为100 mg vs 300 mg、100 mg vs 600 mg、200 mg vs 300 mg、300 mg vs 300 mg。采用同样方法,最终得到不同铁钴比例的四种催化剂:Fe1-Co1-N/C、Fe1-Co2-N/C、Fe2-Co1-N/C、Fe3-Co1-N/C。
1.3 Fe1-Co1-Pt-N/C的制备
将13.81 mg四氯化铂溶于50 mL水中,然后在搅拌下将100 mg Fe1-Co1-N/C纳米片粉末加到上述溶液中,在50℃水浴中加热并继续搅拌4小时。之后过滤并用水洗涤,再加水进行离心分离,所得固体转移到70℃烘箱中干燥。样品烘干后转移到管式炉中,在氮气气氛中以4℃/min的升温速率,分别加热到500℃、550℃、600℃,保持该温度3个小时,得到氮掺杂碳纳米片负载的铂-铁-钴复合材料(Fe1-Co1-Pt-N/C 500,Fe1-Co1-Pt-N/C 550,Fe1-Co1-Pt-N/C 600)。
催化剂制备过程如图1所示。
![]() 图 1 负载铂铁钴三金属合金的氮掺杂碳纳米片(Fe1-Co1-Pt-N/C)催化剂制备流程图Figure 1. Schematic diagram for the synthesis of Pt-Fe-Co loaded nitrogen-doped carbon nanosheet composites (Fe1-Co1-Pt-N/C) catalysts
图 1 负载铂铁钴三金属合金的氮掺杂碳纳米片(Fe1-Co1-Pt-N/C)催化剂制备流程图Figure 1. Schematic diagram for the synthesis of Pt-Fe-Co loaded nitrogen-doped carbon nanosheet composites (Fe1-Co1-Pt-N/C) catalysts2. 表征与测试
利用发射扫描电子显微镜(SEM, FEI Inspect F50, FSEM)、Mapping (EDAX super octane)和透射电镜(TEM, Zeiss Merlin compact-61-78)对催化剂的微观结构和形貌进行表征;利用结合能谱(EDS,Super octane)和X射线光电子光谱(XPS, Thermo Fisher Scientific K-Alpha)对样品元素组成与成键状态进行表征;金属铂的含量在电子耦合等离子体发射光谱仪(ICP-OES730)上测定;采用Bruker D8 ADVANCE A25X对样品进行X射线衍射(XRD)分析;Brunauer - emmet - teller (BET)测试在自动物理吸附仪上进行,模型为3H-2000PM2 Bass。
采用三电极体系,在电化学工作站(AutoLab PGSTAT30/FRA)上测定了催化剂的ORR性能。其中,以涂有催化剂油墨膜的玻碳(GC)、Pt箔和装有饱和KCl溶液的Ag/AgCl电极分别作为工作电极、辅助电极和参比电极。利用方程(ERHE/V = EAg/AgCl+0.197+0.0591×pH)将所有电位转化为相对可逆氢电极(RHE)的电位。工作电极制备过程为:将5 mg催化剂粉末、950 μL乙醇和50 μL Nafion溶液(5wt%,杜邦)混合,超声处理60 min,使混合物形成墨水状的均匀分散液。然后,用微管在预先用Al2O3粉末抛光处理的玻碳电极(0.071 cm2)表面涂上20 μL的催化剂墨水,并让它在室温下自然干燥,得到工作电极。对于所制备的催化剂,在GC上的负载量控制在1.41 mg·cm−2。在饱和氧的0.1 mol·L−1 HClO4溶液中进行了氧还原反应(ORR)电催化活性测试。为了进行比较,采用同样步骤制备了Pt/C (40wt%, Johnson Matthey Corp.)催化剂工作电极,负载量为0.7 mg·cm−2。
通过Koutecky-Levich方程计算ORR过程中传递的电子数,如下所示:
1j=1jL+1jK=1Bω1/2+1jK (1) 其中:j、jL和jK分别为实测电流密度、极限扩散电流密度和动力学电流密度;B为Levich曲线斜率;ω为电极转速。
应用下式所示的B计算n:
B=0.2nFCO2DO22/3v1/6 (2) 其中:n和F分别为电子的数目和法拉第常数(96485 C·mol−1);
CO2 是室温下在电解液中的饱和氧气的浓度(1.2×10−6 mol·cm−3);DO2 是氧气在水中的扩散系数(1.93×10−5 cm2·s−1); ν是室温下溶液的运动黏度 (0.01 cm2·s−1)[18]。3. 结果与讨论
3.1 样品表征
扫描电子显微镜(SEM)和透射电子显微镜(TEM)观察到的样品表面形态以及金属纳米颗粒的外观和分布如图2所示,分别是Fe1-Co1-N/C、Fe1-Co1-Pt-N/C 500、Fe1-Co1-Pt-N/C 550和Fe1-Co1-Pt-N/C 600的SEM形貌图,可以看到所有样品的形貌都是皱褶状的纳米片,催化剂Fe1-Co1-N/C(图2(a))的纳米片表面负载很多明显的金属纳米颗粒,而加入少量铂之后的三种催化剂(图2(b)~2(d))表面并没有明显的金属纳米颗粒,是由于铂的加入促进了碳材料的石墨化,石墨碳壳紧紧地包裹着金属纳米颗粒,以至于遮盖了大量的金属纳米颗粒,从而阻碍了它们的检测[19]。
![]() 图 2 Fe1-Co1-N/C (a)、Fe1-Co1-Pt-N/C 500 (b)、Fe1-Co1-Pt-N/C 550 (c)和Fe1-Co1-Pt-N/C 600 (d)催化剂的SEM图像Figure 2. SEM images of the catalysts Fe1-Co1-N/C (a), Fe1-Co1-Pt-N/C 500 (b), Fe1-Co1-Pt-N/C 550 (c) and Fe1-Co1-Pt-N/C 600 (d)
图 2 Fe1-Co1-N/C (a)、Fe1-Co1-Pt-N/C 500 (b)、Fe1-Co1-Pt-N/C 550 (c)和Fe1-Co1-Pt-N/C 600 (d)催化剂的SEM图像Figure 2. SEM images of the catalysts Fe1-Co1-N/C (a), Fe1-Co1-Pt-N/C 500 (b), Fe1-Co1-Pt-N/C 550 (c) and Fe1-Co1-Pt-N/C 600 (d)进一步从图3(a)所示中的TEM图像看出,催化剂Fe1-Co1-Pt-N/C 550同样是由具有大量皱褶痕的纳米片构成,表面上可以看到均匀分散的大小不同的金属纳米颗粒。图3(b)和图3(c)是催化剂Fe1-Co1-Pt-N/C 550的高倍透射电镜图(HRTEM),可以看到金属颗粒具有大量连续的晶格条纹。从图3(b)中看出,晶格条纹间距为0.221 nm对应的是Pt(111)的面心立方晶面[20],间距为0.175 nm的晶格条纹对应的是Fe或Co的(200)晶面[21]。除此之外,从图3(c)看到晶格条纹间距为0.197 nm的纳米颗粒,对应的是Pt(200)的晶面[22]。由此可见,TEM测试结果对应XRD(图4(a))的表征结果。图3(d)~3(h)是催化剂Fe1-Co1-Pt-N/C 550里各元素的映射图像,可以看到C、N、Pt、Fe、Co五种原子均匀分布在纳米材料上。
![]() 图 3 Fe1-Co1-Pt-N/C 550催化剂的TEM图像(a)和HRTEM图像((b),(c)); Fe1-Co1-Pt-N/C 550催化剂的C(d)、 N (e)、Pt (f)、Fe (g)和Co (h)元素的映射图像Figure 3. TEM image (a) and HRTEM images ((b),(c)) of Fe1-Co1-Pt-N/C 550 catalysts; Mapping images of C (d), N (e), Pt (f), Fe (g) and Co (h) of Fe1-Co1-Pt-N/C 550 catalyst
图 3 Fe1-Co1-Pt-N/C 550催化剂的TEM图像(a)和HRTEM图像((b),(c)); Fe1-Co1-Pt-N/C 550催化剂的C(d)、 N (e)、Pt (f)、Fe (g)和Co (h)元素的映射图像Figure 3. TEM image (a) and HRTEM images ((b),(c)) of Fe1-Co1-Pt-N/C 550 catalysts; Mapping images of C (d), N (e), Pt (f), Fe (g) and Co (h) of Fe1-Co1-Pt-N/C 550 catalyst![]() 图 4 Fe1-Co1-N/C and Fe1-Co1-Pt-N/C催化剂的XRD图谱(a)和EDS能谱图(b)Figure 4. XRD patterns (a) and EDS spectra (b) of the Fe1-Co1-N/C and Fe1-Co1-Pt-N/C catalysts
图 4 Fe1-Co1-N/C and Fe1-Co1-Pt-N/C催化剂的XRD图谱(a)和EDS能谱图(b)Figure 4. XRD patterns (a) and EDS spectra (b) of the Fe1-Co1-N/C and Fe1-Co1-Pt-N/C catalysts通过X-射线衍射(XRD)和扫描电子显微镜能谱(EDS)来了解样品的元素组成并分析材料相组成。从图4(a)所示的催化剂XRD谱看出,催化剂Fe1-Co1-Pt-N/C 550和Fe1-Co1-Pt-N/C 600在2θ=26.6°处出现了石墨碳衍射峰对应的(002)晶面[23],而催化剂Fe1-Co1-N/C和Fe1-Co1-Pt-N/C 500的石墨碳衍射峰负移到2θ=21.3°处,反映了界面距离的增大,表明过渡金属(Fe、Co)和Pt掺杂有利于增大石墨烯片层之间的距离并减小石墨烯片层之间的相互作用,从而抑制石墨烯的团聚;同时较弱的宽衍射峰表明金属纳米颗粒掺杂导致石墨烯的结晶取向变差。催化剂Fe1-Co1-N/C在2θ=43.38°处出现明显的衍射峰,对照Fe (PDF#06-0696)和Co (PDF#05-0727)的标准卡片,这个峰位于Fe(2θ=44.67°)和Co(2θ=41.68°)的标准峰之间,表明形成部分铁钴合金,这可以归属于铁或钴面心立方结构的(111)点阵面[19]。除此之外,Fe1-Co1-Pt-N/C 500、Fe1-Co1-Pt-N/C 550和Fe1-Co1-Pt-N/C 600在2θ=40.0°和46.5°处出现两个明显的衍射峰,根据Pt(PDF#04-0802)的标准卡片,对应Pt位于2θ=39.76°和2θ=46.24°处的标准峰,归属于Pt晶格面心立方的(111)点阵面和(200)点阵面,与纯Pt相比,这两个峰稍微向更高的角度偏移,说明形成Pt-Fe复合物或Pt-Co复合物[24-26];除此之外,2θ=46.5°位于Fe(2θ=44.67°)和Co(2θ=41.68°)的标准峰之间,也表明形成部分铁钴合金,对应于铁或钴的(200)晶格面心立方结构。 因此,XRD表征结果与上述TEM(图3(b)~3(c))分析结果一致。EDS光谱(图4(b))进一步表明四种催化剂Fe-Co-N/C 由C、N、O、Fe和Co元素组成,三种Fe1-Co1-Pt-N/C 催化剂的EDS光谱表明已经成功掺杂Pt原子。样品中C, N, O, Fe和Co元素的含量EDS测定,Pt含量采用ICP测定,结果列于表1中。结果表明,催化剂Fe1-Co1-Pt-N/C 500、Fe1-Co1-Pt-N/C 550和Fe1-Co1-Pt-N/C 600 中Pt的质量分数分别是2.36wt%、3.58wt%和2.69wt%。
表 1 催化剂不同元素的含量Table 1. Contents of different elements in catalystwt% Sample Percentage by mass/wt% EDS data ICP data C N O Fe Co Pt Fe1-Co1-N/C 58.25 39.14 — 0.13 2.48 — Fe1-Co2-N/C 62.24 33.04 — 0.2 7.23 — Fe2-Co1-N/C 56.60 37.78 — 0.29 5.33 — Fe3-Co1-N/C 59.61 32.92 — 0.24 4.52 — Fe1-Co1-Pt-N/C 500 53.69 29.63 5.48 0.33 5.31 2.36 Fe1-Co1-Pt-N/C 550 53.52 27.30 4.90 0.71 7.14 3.58 Fe1-Co1-Pt-N/C 600 55.24 27.88 4.72 0.55 7.73 2.69 为了探索催化剂的催化性能机制,通过氮气吸脱附测试,得到三种Fe1-Co1-Pt-N/C催化剂的比表面积(BET)以及孔体积。如图5所示,催化剂Fe1-Co1-Pt-N/C 500、Fe1-Co1-Pt-N/C 550和Fe1-Co1-Pt-N/C 600的比表面积分别是59.20 m2·g−1、90.52 m2·g−1和106.12 m2·g−1,对应的孔体积分别是0.3595 cm·g−1、0.3837 cm·g−1和0.4137 cm·g−1。催化剂比表面积数值说明纳米片之间堆叠和团聚现象较多,这和SEM得到的形貌图(图2)结果一致。煅烧温度从500℃升到550℃,催化剂比表面积增大了53%,因此Fe1-Co1-Pt-N/C 500的催化性能不如Fe1-Co1-Pt-N/C 550是由于无序化程度较低,说明催化剂从550℃开始得到较为良好的碳化。Fe1-Co1-Pt-N/C 550和Fe1-Co1-Pt-N/C 600的比表面积只相差16 m2·g−1,而催化剂Fe1-Co1-Pt-N/C 550氧还原催化性能(图7(c))更好,可能是由于样品在550℃下得到了更好的碳化,以及在该温度下生成的金属合金,以及金属-C或金属-N物质更有利于增强电活性。
![]() 图 5
图 5Fe1-Co1-Pt-N/C 500 (a)、Fe1-Co1-Pt-N/C 550 (b)和Fe1-Co1-Pt-N/C 600(c)催化剂吸脱附曲线图以及孔体积(插图) Figure 5. Nitrogen adsorption-desorption isotherms and corresponding pore size distributions (insetting) of Fe1-Co1-Pt-N/C 500 (a), Fe1-Co1-Pt-N/C 550 (b) and Fe1-Co1-Pt-N/C 600 (c)Vad—Volume of adsorption通过X射线光电子能谱(XPS)进一步研究了催化剂Fe1-Co1-N/C 和Fe1-Co1-Pt-N/C 550复合材料的表面化学组成。图6(a)~6(b)是催化剂Fe1-Co1-N/C和Fe1-Co1-Pt-N/C 550的全谱分析,图6(a)显示了催化剂Fe1-Co1-N/C 由C、N、O、Fe和Co元素组成,而催化剂Fe1-Co1-Pt-N/C 550由C、N、O、Fe、Co和Pt元素组成(图6(b))。进一步从图6(c)看出,Pt4f7/2和Pt4f5/2的结合能在73.08 eV和76.04 eV的位置,符合零价铂的Pt4f7/2和Pt4f5/2值,说明合成的复合材料中存在单质铂,这一结果与XRD分析一致。图6(d)~6(h)是催化剂Fe1-Co1-Pt-N/C 550的Fe2p、Co2p、Pt4f、C1s和N1s的分峰光谱。图6(d)显示,Fe2p可以反卷积为三个峰,在结合能710.7 eV对应的是Fe2+,711.1 eV对应的是Fe3+,说明Fe2O3和Fe3O4两种物质的存在,除此之外,在结合能706.8 eV对应的是Fe/Fe-N物质[19],是ORR活性基团的主要来源之一。图6(e)显示,Co2p可以反卷积为三个峰,在结合能781.9 eV、779.9 eV和778.4 eV处对应的是Co3+、Co2+、Co0三类物种[19]。图6(f)显示Pt4f可以反卷积为四个特征峰。结合能72.22 eV和75.68 eV处对应的是Pt0物种,结合能72.94 eV和76.21 eV分别对应的是Pt2+和Pt4+物种[18, 27]。进一步从图6(g)看出,C1s高分辨率XPS能谱显示出284.52 eV、285.49 eV、286.65 eV和288.1 eV四个主峰,分别对应C=C、C—O/C=N、O—C=O和C=O/C—N键[17]。如图6(h)所示,N1s高分辨光谱分别出现了398.4 eV、400.4 eV、401.4 eV三个峰[28],分别对应吡啶-N(36.4%)、吡咯-N(32.2%)和石墨-N(31.3%),这说明经过高温煅烧后,氮元素以更稳定的形式存在,并且吡啶-N和石墨-N的存在进一步证实了杂原子通过原位掺杂的方式掺杂到碳骨架中,而且高含量的吡啶-N可以改变碳原子的电子离域效应,减少电子转移能的损失,从而有效增强电子转移[29]。
![]() 图 6 Fe1-Co1-N/C (a)和Fe1-Co1-Pt-N/C 550 (b)的XPS全谱; (c) Fe1-Co1-Pt-N/C 550的Pt4f的XPS谱; Fe1-Co1-Pt-N/C 550的Fe2p (d)、Co2p (e)、Pt4f (f)、C1s (g)和N1s (h)的XPS分谱Figure 6. Full-range XPS of Fe1-Co1-N/C (a) and Fe1-Co1-Pt-N/C 550 (b); XPS of Pt4f of Fe1-Co1-Pt-N/C 550 (c); XPS spectra of Fe2p (d), Co2p (e), Pt4f (f), C1s (g) and N1s (h) in Fe1-Co1-Pt-N/C 550 catalyst
图 6 Fe1-Co1-N/C (a)和Fe1-Co1-Pt-N/C 550 (b)的XPS全谱; (c) Fe1-Co1-Pt-N/C 550的Pt4f的XPS谱; Fe1-Co1-Pt-N/C 550的Fe2p (d)、Co2p (e)、Pt4f (f)、C1s (g)和N1s (h)的XPS分谱Figure 6. Full-range XPS of Fe1-Co1-N/C (a) and Fe1-Co1-Pt-N/C 550 (b); XPS of Pt4f of Fe1-Co1-Pt-N/C 550 (c); XPS spectra of Fe2p (d), Co2p (e), Pt4f (f), C1s (g) and N1s (h) in Fe1-Co1-Pt-N/C 550 catalyst![]() 图 7 扫描速率为50 mV·s−1时,所有催化剂分别在O2和N2饱和的0.1 mol·L−1 HClO4溶液中的CV曲线(a);催化剂Fe-Co-N/C (b)、催化剂Fe1−Co1−Pt-N/C和Pt/C (c)在0.1 mol·L−1 HClO4溶液中1600 r/min转速下的LSV曲线;(d) 催化剂Fe1−Co1−Pt-N/C 550在不同转速下的LSV曲线;(e) ORR起始电位比较;(f) ORR极限电流比较Figure 7. At scanning rate of 5 mV·s−1, CV curves (a) of all catalysts in 0.1 mol·L−1 HClO4 solution saturated with O2 and N2; Comparison of LSV curves of Fe-Co-N/C catalysts (b), Fe1−Co1−Pt-N/C catalyst and catalyst Pt/C (c) at 1600 r/min in 0.1 mol·L−1 HClO4 solution; (d) LSV curves of the Fe1−Co1−Pt-N/C 550 catalyst at different rotation speed; (e) ORR onset potential comparison; (f) ORR limiting current comparisonE—Electric potential; j—Limiting diffusion current; w—Rotate speed; n—Electron transfer number
图 7 扫描速率为50 mV·s−1时,所有催化剂分别在O2和N2饱和的0.1 mol·L−1 HClO4溶液中的CV曲线(a);催化剂Fe-Co-N/C (b)、催化剂Fe1−Co1−Pt-N/C和Pt/C (c)在0.1 mol·L−1 HClO4溶液中1600 r/min转速下的LSV曲线;(d) 催化剂Fe1−Co1−Pt-N/C 550在不同转速下的LSV曲线;(e) ORR起始电位比较;(f) ORR极限电流比较Figure 7. At scanning rate of 5 mV·s−1, CV curves (a) of all catalysts in 0.1 mol·L−1 HClO4 solution saturated with O2 and N2; Comparison of LSV curves of Fe-Co-N/C catalysts (b), Fe1−Co1−Pt-N/C catalyst and catalyst Pt/C (c) at 1600 r/min in 0.1 mol·L−1 HClO4 solution; (d) LSV curves of the Fe1−Co1−Pt-N/C 550 catalyst at different rotation speed; (e) ORR onset potential comparison; (f) ORR limiting current comparisonE—Electric potential; j—Limiting diffusion current; w—Rotate speed; n—Electron transfer number3.2 催化剂电化学测试
用循环伏安法(CV)测定了所有催化剂在酸性溶液中对ORR的电催化活性。在N2和O2饱和的0.1 mol·L−1 HClO4溶液中,扫描速度为50 mV·s−1时,样品的CV曲线如图7(a)和图7(b)的插图所示。在N2饱和的电解液中没有观察到明显的阴极峰,而在O2饱和时,所有样品的阴极电流密度都有增加,并出现明显的阴极峰,说明其对氧还原具有电催化活性。根据光滑石墨表面的双电层比电容(20 μF·cm−2)[30],从N2饱和下的循环伏安曲线(图7(a)),得到催化剂Fe1-Co1-N/C、Fe1-Co1-Pt-N/C 500、Fe1-Co1-Pt-N/C 550和Fe1-Co1-Pt-N/C 600的电化学活性表面积分别为270、580、425和400 cm−2。利用旋转圆盘电极(RDE)进一步评估了催化剂的ORR性能,图7(b)和图7(c)是所有催化剂在0.1 mol·L−1 HClO4溶液中的线性扫描曲线(LSV))。图7(b)比较了不同铁钴比例催化剂的LSV曲线,可以看到催化剂Fe1-Co1-N/C、Fe2-Co1-N/C、Fe3-Co1-N/C、Fe1-Co2-N/C对应的ORR起始电位分别是0.82、0.77、0.79、0.71 V,在0.3 V时对应的极限扩散电流分别是4.97、3.74、3.89和3.24 mA·cm−2,由此可见,催化剂Fe1-Co1-N/C 在0.1 mol·L−1 HClO4溶液中的起始电位和极限扩散电流最大,表明其氧还原性能最优。通过在Fe1-Co1-N/C 中引入少量铂,使形成的催化剂Fe1-Co1-Pt-N/C的ORR活性进一步提升。如图7(c)所示,可以看到催化剂Fe1-Co1-N/C和含低载量铂的三种催化剂Fe1-Co1-Pt-N/C 500、Fe1-Co1-Pt-N/C 550和Fe1-Co1-Pt-N/C 600在0.1 mol·L−1 HClO4溶液中在1600 r/min转速下对ORR的起始电位分别是0.82、0.87、0.96和0.94 V,在电位0.2 V时对应的极限扩散电流分别是5.10、6.27、6.88和5.91 mA·cm−2。同时,在0.44~0.73 V的电位范围内,ORR电流密度呈快速增加,电位负移,但相比于Fe1-Co1-N/C,其它含铂样品的电位负移程度明显下降,说明它们的ORR半波电位提前,极大改善了ORR动力学过程。由此可见,加入少量Pt之后,与催化剂Fe1-Co1-N/C相比,催化剂Fe1-Co1-Pt-N/C的氧还原性能得到明显改善。其中,催化剂Fe1-Co1-Pt-N/C 550表现出最好的氧还原活性,其起始电位和扩散电流甚至超过了Pt/C(0.94 V,5.91 mA·cm−2),具体数据对比见图7(e)~7(f)。图7(d)是催化剂Fe1-Co1-Pt-N/C 550在不同转速下的LSV曲线,可以看到,由于氧的扩散层厚度降低,极限扩散电流密度随转速的增加而增大;同时,根据催化剂Levich曲线的斜率(B),计算出催化剂在酸性介质中的ORR电子转移数(n)在3.6~4.0之间,属于四电子转移过程,结果如图7(d)插图所示。由于ORR的四电子转移过程比双电子转移过程具有更高的输出电压,因此认为ORR的四电子转移过程更利于氧还原阴极反应[18, 28]。除此之外,对催化剂Fe1-Co1-N/C 和Fe1-Co1-Pt-N/C 550进行了氧还原的活性稳定性测试,结果如图8(a)~8(b)所示。在1600 r/min下对Fe1-Co1-N/C 进行了120次循环测试,催化剂的起始电位没有明显改变,只有电流产生了5%的偏移,而Fe1-Co1-Pt-N/C 550进行120次循环之后,起始电位和极限扩散电流几乎没有发生变化,曲线基本保持一致,表明催化剂Fe1-Co1-N/C和Fe1-Co1-Pt-N/C 550具备良好的电催化稳定性。结果表明,本工作制备的低载铂催化剂具有优异的氧还原催化性能,具有较好的应用前景。
![]() 图 8 扫描速率为10 mV·s−1时,催化剂Fe1-Co1-N/C (a)和Fe1-Co1-Pt-N/C 550 (b)在1 600 r/min下连续120次循环测试的LSV曲线Figure 8. LSV curves of the Fe1-Co1-N/C (a) and Fe1-Co1-Pt-N/C 550 (b) at 1 600 r/min and 10 mV·s−1 for 120 successive cycles
图 8 扫描速率为10 mV·s−1时,催化剂Fe1-Co1-N/C (a)和Fe1-Co1-Pt-N/C 550 (b)在1 600 r/min下连续120次循环测试的LSV曲线Figure 8. LSV curves of the Fe1-Co1-N/C (a) and Fe1-Co1-Pt-N/C 550 (b) at 1 600 r/min and 10 mV·s−1 for 120 successive cycles4. 总 结
(1) 合成了负载铁钴合金的氮掺杂碳纳米片(Fe1-Co1-N/C)以及负载铂铁钴三金属合金的氮掺杂碳纳米片复合物(Fe1-Co1-Pt-N/C)催化剂,铂质量分数在2.36wt% ~ 3.58wt%之间。
(2) 酸性溶液中Fe1-Co1-N/C本身对氧还原反应 (ORR) 具有较好的电催化活性;虽然Fe1-Co1-Pt-N/C的铂负载量低,但其ORR起始电位和极限电流密度均达到或超过Pt/C,单位质量铂表现出极高的ORR电流,并且具有优异的活性稳定性。
(3) Fe1-Co1-N/C催化剂中较高的吡啶氮和石墨氮含量、金属之间形成的合金和协同效应,以及金属-N等活性基团的存在,是导致催化剂优异ORR电活性的主要原因。
-
![]()
图 4 CZS-0.3 (a)、CeO2 (b)、10%CCZS-0.3 (c)的SEM图像
Figure 4. SEM images of CZS-0.3 (a), CeO2 (b), 10%CCZS-0.3 (c)
![]()
图 5 ((a)~(c)) 10%CCZS-0.3的HR-TEM图像;(d) 10%CCZS-0.3的EDS mapping
Figure 5. ((a)-(c)) HR-TEM images of 10%CCZS-0.3; (d) EDS mapping of 10%CCZS-0.3
d—Interplanar spacing
![]()
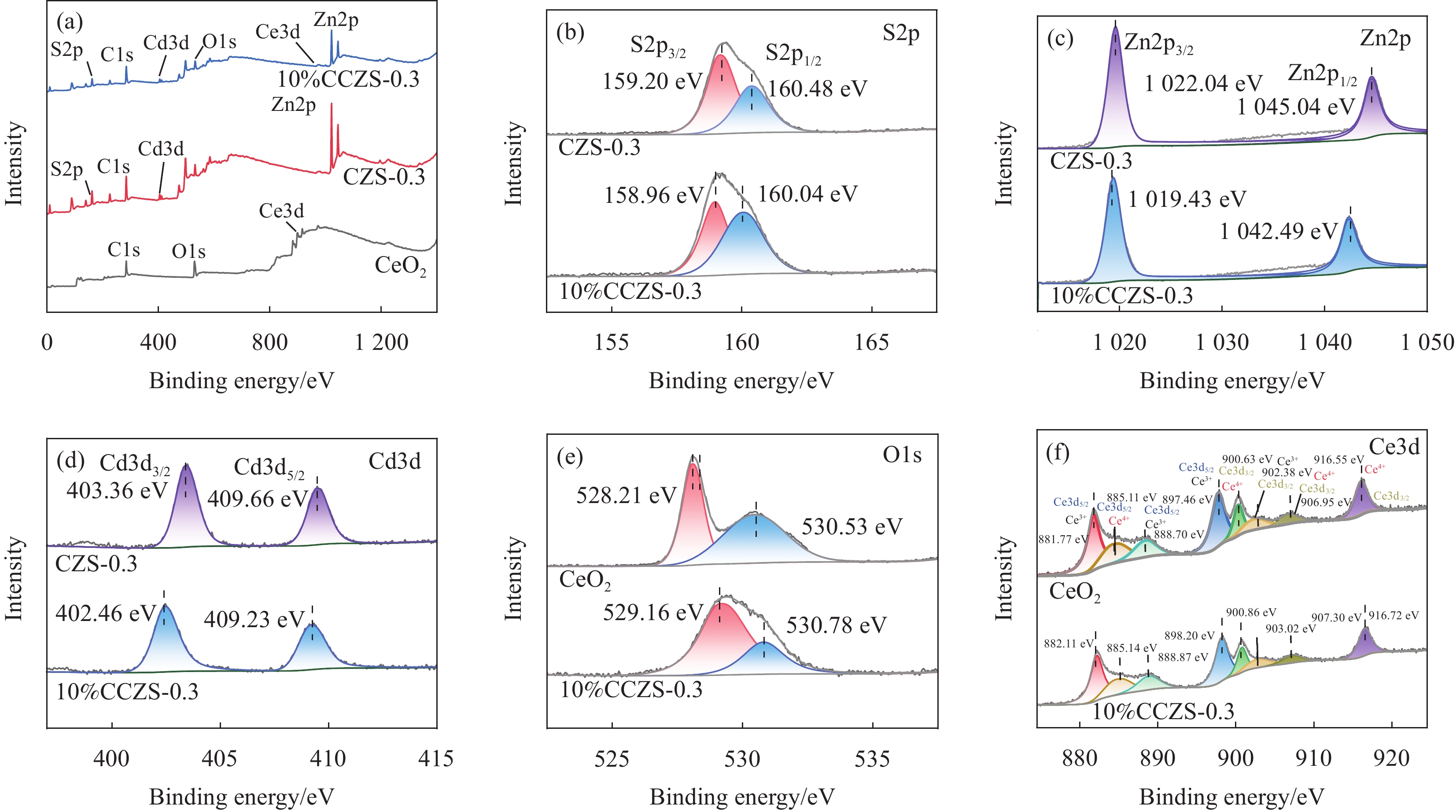
图 6 (a)样品的XPS全谱;样品的XPS:(b) S2p;(c) Zn2p;(d) Cd3d;(e) O1s;(f) Ce3d
Figure 6. (a) XPS full spectrum of the sample; XPS of the sample: (b) S2p; (c) Zn2p; (d) Cd3d; (e) O1s; (f) Ce3d
![]()
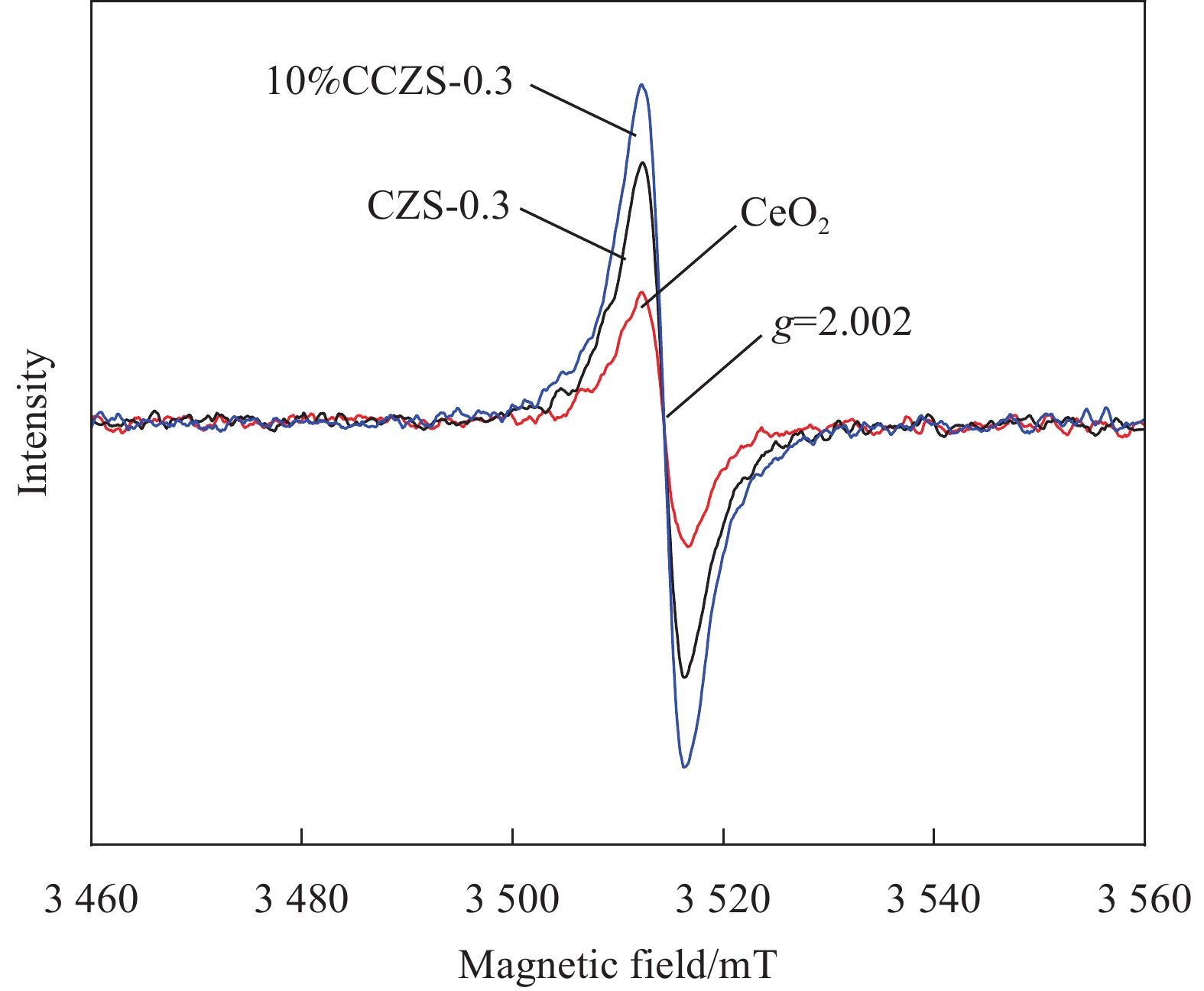
图 7 CeO2、CZS-0.3、10%CCZS-0.3的EPR图谱
Figure 7. EPR spectra of CeO2, CZS-0.3, 10%CCZS-0.3
g—No dimensional factor
![]()
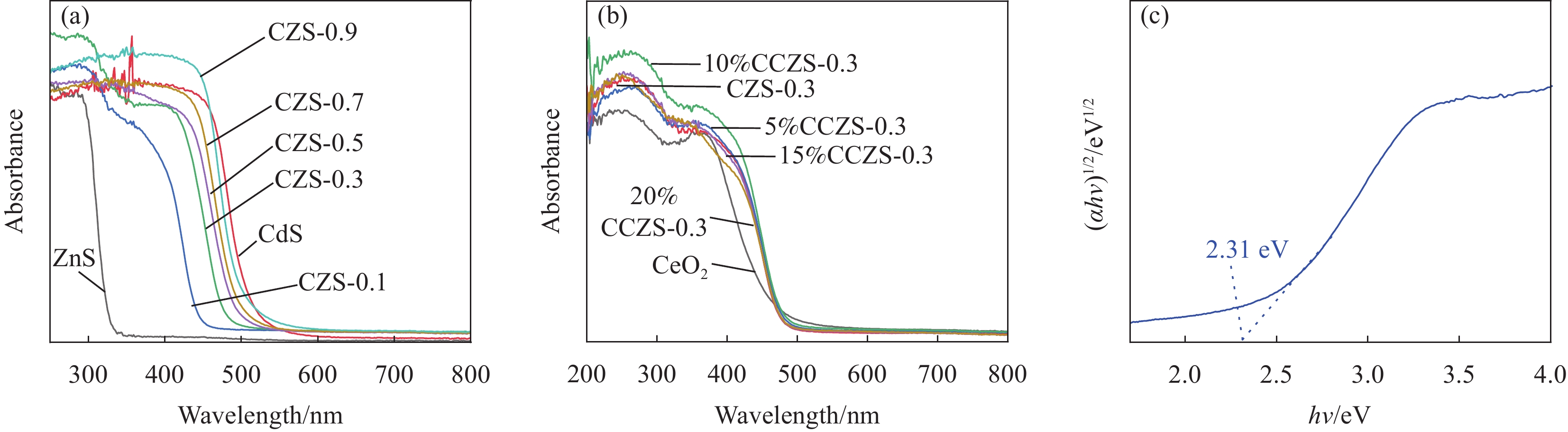
图 8 (a) CZS-X的UV-Vis DRS谱图;(b) CeO2、CZS-0.3、10%CCZS-0.3的UV-Vis DRS谱图;(c) CeO2的带隙
Figure 8. (a) UV-Vis DRS of CZS-X; (b) UV-Vis DRS of CeO2, CZS-0.3 and 10%CCZS-0.3; (c) Band gap of CeO2
α—Absorption coefficient; h—Planck constant; v—Incident light frequency
![]()
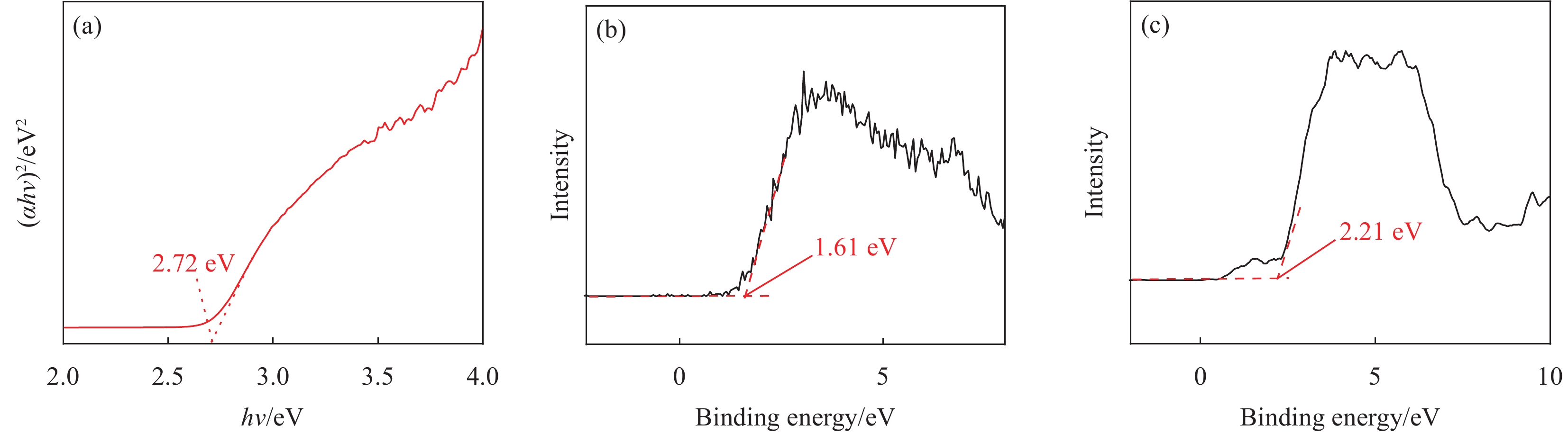
图 9 (a) CZS-0.3的带隙;(b) CeO2的XPS价带谱;(c) CZS-0.3的XPS价带谱
Figure 9. (a) Band gap of CZS-0.3; (b) Valence-band spectrum of CeO2; (c) Valence-band spectrum of CZS-0.3
![]()
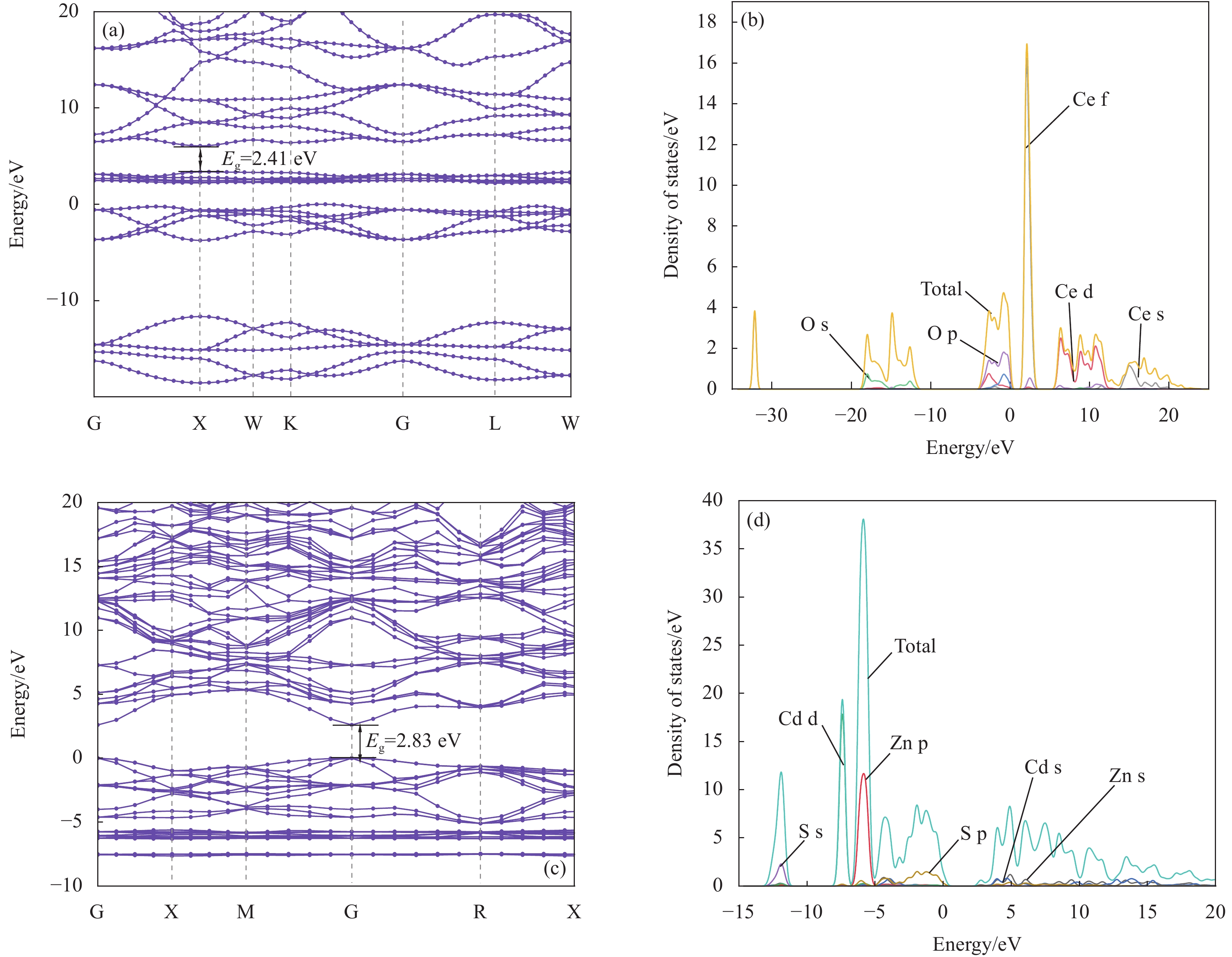
图 10 (a) CeO2的能带结构;(b) CeO2的态密度;(c) CZS-0.3的能带结构;(d) CZS-0.3的态密度
Figure 10. (a) Band structure of CeO2; (b) Density of states of CeO2; (c) Band structure of CZS-0.3; (d) Density of states of CZS-0.3
Eg—Band gap
![]()
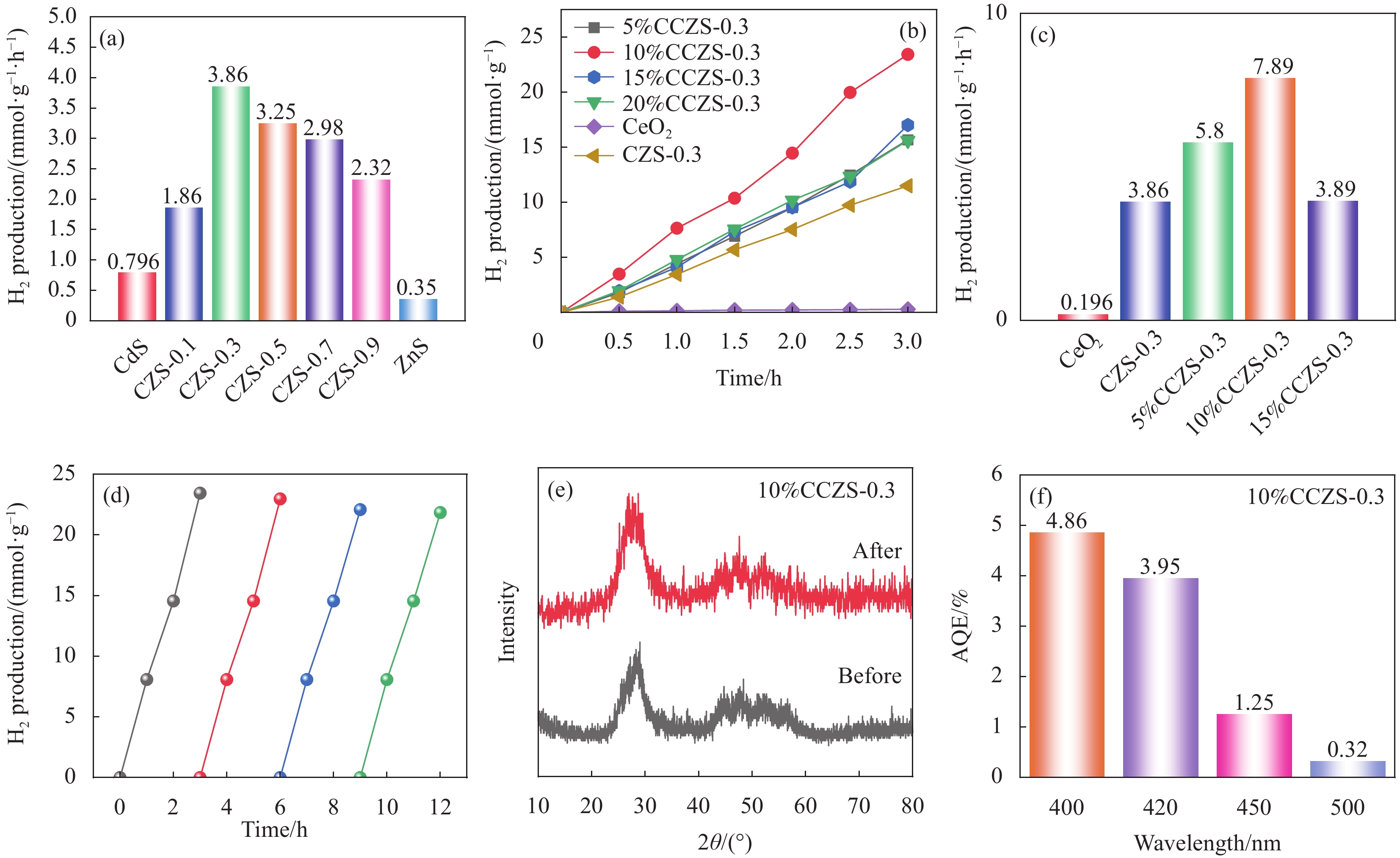
图 11 (a) CZS-X的光催化产氢性能;(b) CeO2、y%CCZS-0.3的光催化产氢性能;(c) y%CCZS-0.3的光催化产氢性能;(d) 10%CCZS-0.3的产氢稳定性试验;(e) 10%CCZS-0.3反应前后XRD图谱;(f) 10%CCZS-0.3的量子效率(AQE)
Figure 11. (a) Photocatalytic hydrogen production of CZS-X; (b) Photocatalytic hydrogen production of CeO2 and y%CCZS-0.3; (c) Photocatalytic hydrogen production of y%CCZS-0.3; (d) Hydrogen production stability of 10%CCZS-0.3; (e) XRD patterns of before and after reaction of 10%CCZS-0.3; (f) Apparent quantum efficiency (AEQ) of 10%CCZS-0.3
![]()
图 12 CeO2、CZS-0.3、10%CCZS-0.3的瞬态光电流谱(a)、电化学阻抗谱(b)、光致发光(PL)光谱(c);((d), (e)) CZS-0.3和CeO2的Mott-Schottky曲线;(i) CZS-0.3和CeO2的能带结构示意图
Figure 12. Transient photocurrent responses (a), EIS spectra (b), PL spectra (c) of CeO2, CZS-0.3 and 10%CCZS-0.3; ((d), (e)) Mott schottky curves of CZS-0.3 and CeO2; (i) Band structure of CZS-0.3 and CeO2
Z'—Real part of impedance; Z"—Imaginary part of impedance; C−2—Inverse of the square of the capacitance; CB—Conduction band; VB—Valence band
![]()
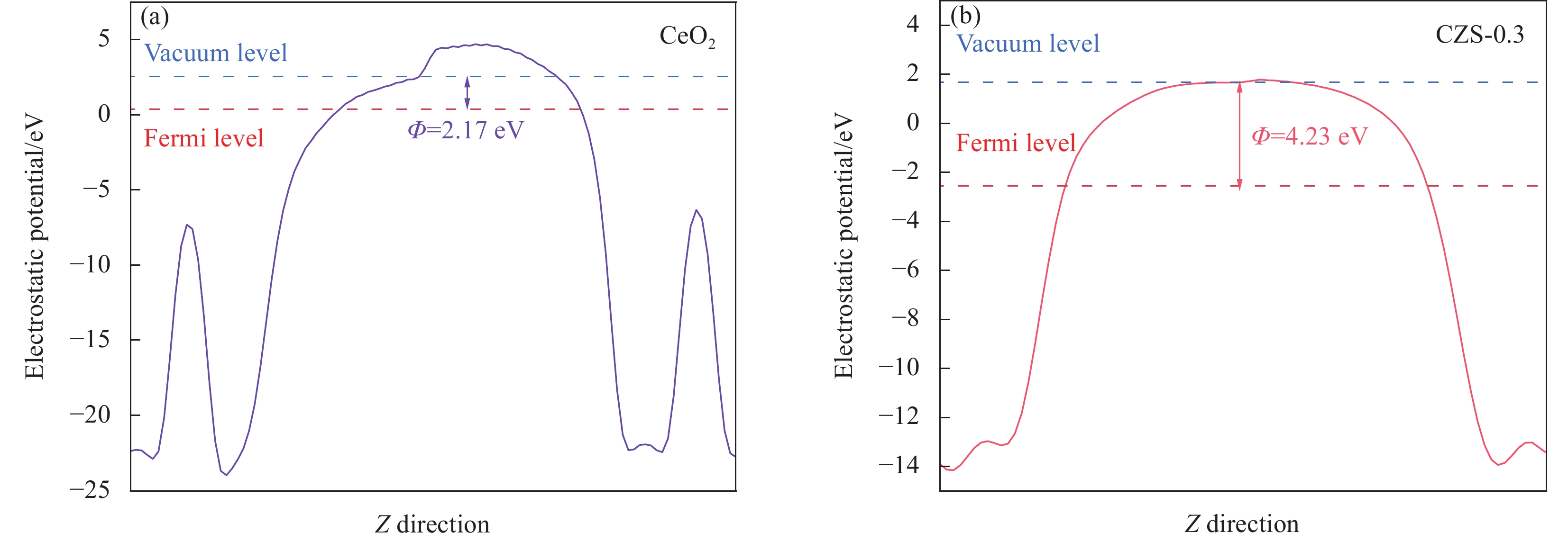
图 13 CeO2 (a)和CZS-0.3 (b)的功函数
Figure 13. Work function calculation of CeO2 (a) and CZS-0.3 (b)
Φ—Work function
![]()
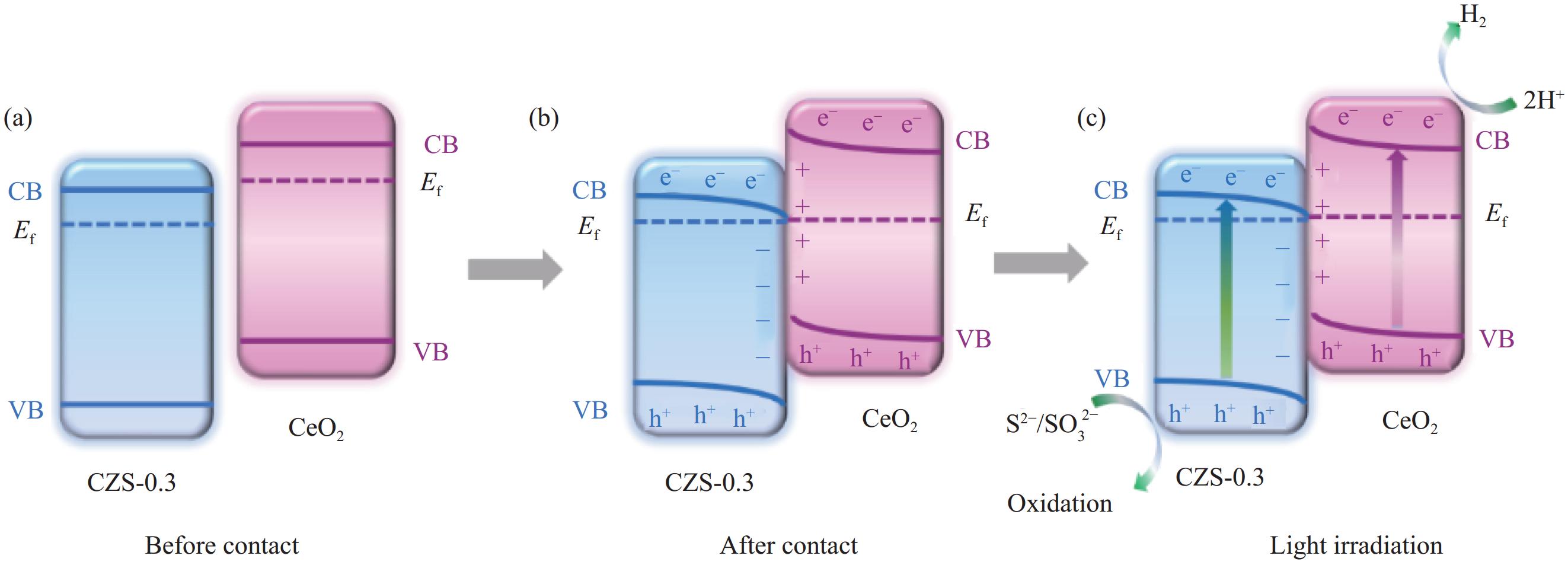
图 14 光催化产氢机制示意图:(a) CeO2与CZS-0.3接触前;(b) CeO2与CZS-0.3形成异质结后;(c)光照下10%CCZS-0.3异质结光生电荷转移途径
Figure 14. Schematic diagram of photocatalytic hydrogen production mechanism: (a) Before forming heterojunction; (b) After forming heterojunction; (c) Photogenerated charge transfer pathway of 10%CCZS-0.3 heterojunction under light
Ef—Fermi level
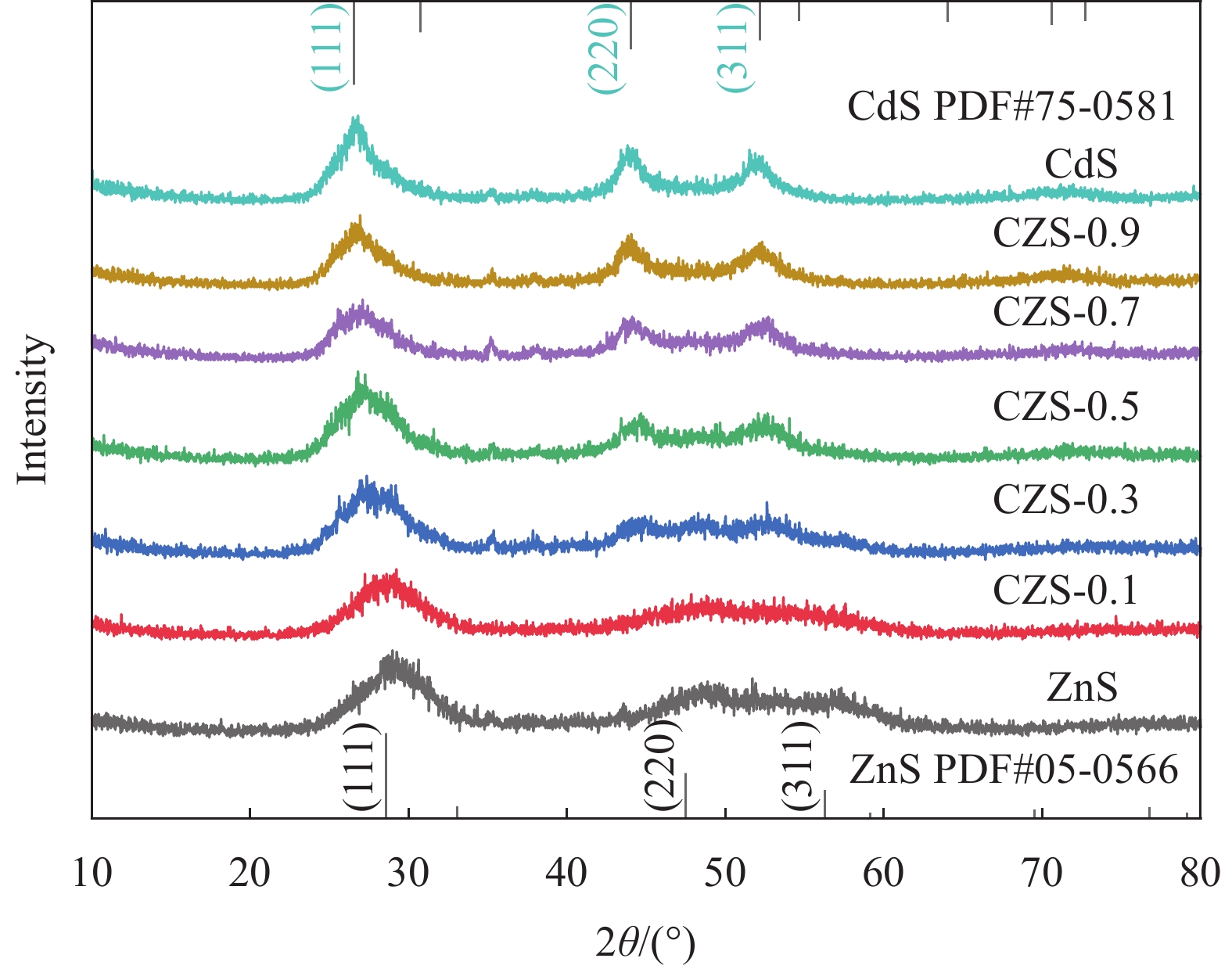
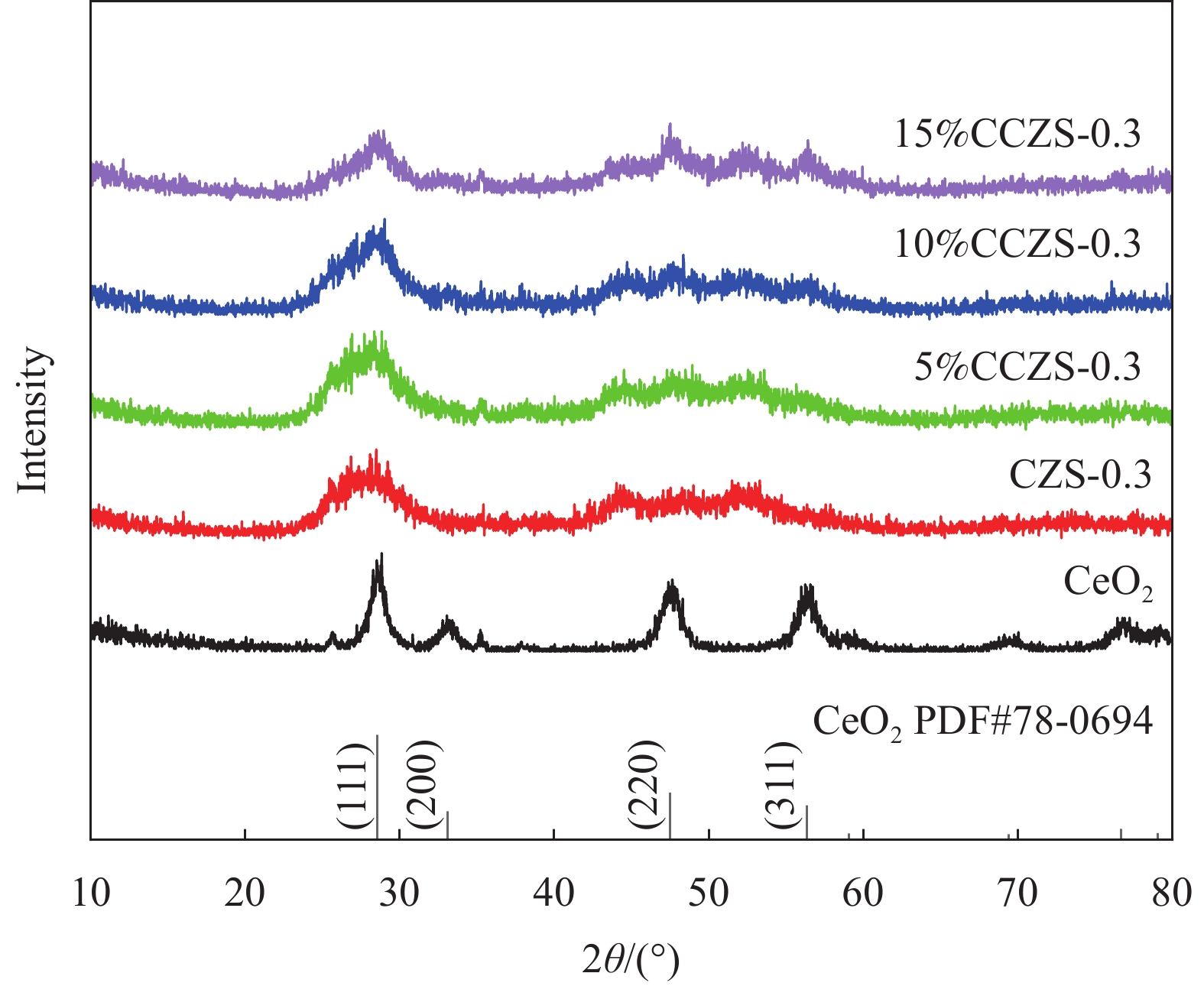
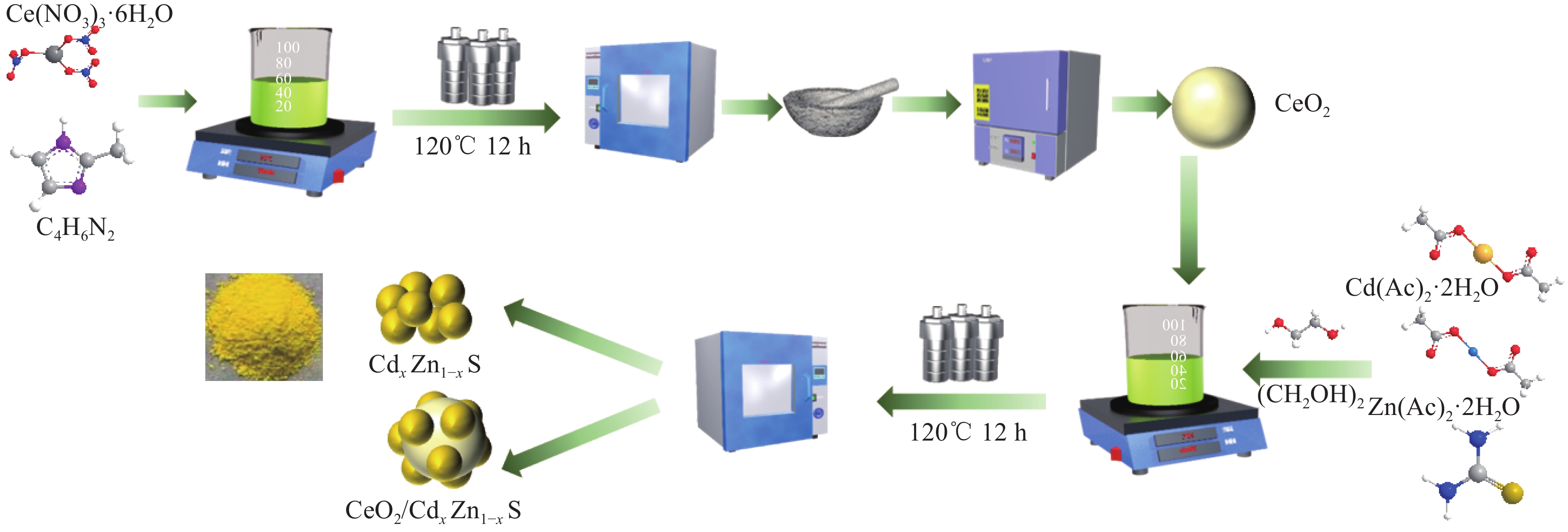
表 1 CdxZn1−xS (CZS-X)和CeO2/CZS (y%CCZS)样品命名
Table 1 Naming of CdxZn1−xS (CZS-X) and CeO2/CZS (y%CCZS)
Sample Mole ratio of
Cd2+∶Zn2+Sample CeO2 content/g CdS 0∶1 CeO2 0 CZX-0.1 0.1∶0.9 5%CCZX-0.3 0.0352 CZX-0.3 0.3∶0.7 10%CCZX-0.3 0.0744 CZX-0.5 0.5∶0.5 15%CCZX-0.3 0.1181 CZX-0.7 0.7∶0.3 20%CCZX-0.3 0.1673 CZX-0.9 0.9∶0.1 ZnS 1∶0  下载: 导出CSV
下载: 导出CSV
-
[1] ZENG R, CHENG C, XING F, et al. Dual vacancies induced local polarization electric field for high-performance photocatalytic H2 production[J]. Applied Catalysis B: Environmental, 2022, 316: 121680. DOI: 10.1016/j.apcatb.2022.121680
[2] JIANG K B, HUANG W Q, SONG T T, et al. Photobreeding heterojunction on semiconductor materials for enhanced photocatalysis[J]. Advanced Functional Materials, 2023, 33(43): 2304351. DOI: 10.1002/adfm.202304351
[3] LUAN X, YU Z, ZI J, et al. Photogenerated defect-transit dual S-scheme charge separation for highly efficient hydrogen production[J]. Advanced Functional Materials, 2023, 33(42): 2304259. DOI: 10.1002/adfm.202304259
[4] JADHAV S R, MOHITE S V, LEE C, et al. In-situ synthesized oxygen vacancy filled ZnS/Vo-ZnO heterojunction photocatalysts for efficient H2 production[J]. Sustainable Materials and Technologies, 2023, 38: e00731. DOI: 10.1016/j.susmat.2023.e00731
[5] YU L, LI X, DUAN L, et al. Oxygen vacancies-induced dendritic SrTiO3/CdS p-n heterostructures photocatalyst for ultrahigh hydrogen evolution[J]. Solar RRL, 2023, 7(14): 2300259. DOI: 10.1002/solr.202300259
[6] AN S, ZHANG L, DING X, et al. A general strategy for the enhanced H2 production performance of CdS/noble metal sulfide nanorods photocatalysts by cation exchange[J]. Journal of Colloid and Interface Science, 2024, 664: 848-856. DOI: 10.1016/j.jcis.2024.03.087
[7] SONG Y, ZHENG X, YANG Y, et al. Heterojunction engineering of multinary metal sulfide-based photocatalysts for efficient photocatalytic hydrogen evolution[J]. Advanced Materials, 2024, 36(11): 2305835. DOI: 10.1002/adma.202305835
[8] WU P, LIU H, XIE Z, et al. Excellent charge separation of NCQDs/ZnS nanocomposites for the promotion of photocatalytic H2 evolution[J]. ACS Applied Materials & Interfaces, 2024, 16(13): 16601-16611.
[9] XING J, WANG Y, SHI G, et al. Defective Cd0.3Zn0.7S/1T-2H MoS2 Z-scheme heterojunctions: Rational design with efficient charge transfer for enhanced photocatalytic H2 generation[J]. Journal of Alloys and Compounds, 2024, 988: 174302. DOI: 10.1016/j.jallcom.2024.174302
[10] MING Y, CHENG Z, SHI S, et al. Nanoarchitectonics toward full coverage of CdZnS nanospheres by layered double hydroxides for enhanced visible-light-driven H2 evolution[J]. Small, 2024, 20(28): 2309750.
[11] YANG L, TIAN Q, WANG X, et al. Interfacial-engineered Co3S4/MnCdS heterostructure for efficient photocatalytic hydrogen evolution[J]. Solar RRL, 2023, 7(17): 2300403. DOI: 10.1002/solr.202300403
[12] ZHANG W, HUANG Z, ZHANG L, et al. Construction of zinc-oxygen double vacancies BiOCl/ZnS Z-scheme heterojunction and photocatalytic degradation of norfloxacin[J]. Journal of Environmental Chemical Engineering, 2023, 11(3): 109979. DOI: 10.1016/j.jece.2023.109979
[13] ZHENG Y, WANG Y, MANSOOR S, et al. Tuning electrons migration of dual S defects mediated MoS2−x/ZnIn2S4−x toward highly efficient photocatalytic hydrogen production[J]. Small, 2024, 20(33): 2311725.
[14] YAN Y Q, WU Y Z, WU Y H, et al. Recent advances of CeO2-based composite materials for photocatalytic applications[J]. ChemSusChem, 2024, 17(14): e202301778.
[15] GAO X, HE H, ZHU W, et al. Continuously flow photothermal catalysis efficiently CO2 reduction over S-scheme 2D/0D Bi5O7I-OVs/Cd0.5Zn0.5S heterojunction with strong interfacial electric field[J]. Small, 2023, 19(12): 2206225.
[16] ZOU X, SUN B, WANG L, et al. Enhanced photocatalytic degradation of tetracycline by SnS2/Bi2MoO6− x heterojunction: Multi-electric field modulation through oxygen vacancies and Z-scheme charge transfer[J]. Chemical Engineering Journal, 2024, 482: 148818. DOI: 10.1016/j.cej.2024.148818
[17] CHEN C, LI Q, WANG F, et al. Dual-vacancies modulation of 1T/2H heterostructured MoS2-CdS nanoflowers via radiolytic radical chemistry for efficient photocatalytic H2 evolution[J]. Journal of Colloid and Interface Science, 2024, 661: 345-357. DOI: 10.1016/j.jcis.2024.01.200
[18] ZHANG C, WANG Y, ZHANG X, et al. Self-nitrogen-doped carbon spheres assisted CeO2 composites as a bifunctional adsorbent/photocatalyst for CO2 photoreduction[J]. Fuel, 2024, 362: 130848. DOI: 10.1016/j.fuel.2023.130848
[19] SHEN C H, CHEN Y, XU X J, et al. Efficient photocatalytic H2 evolution and Cr(VI) reduction under visible light using a novel Z-scheme SnIn4S8/CeO2 heterojunction photocatalysts[J]. Journal of Hazardous Materials, 2021, 416: 126217. DOI: 10.1016/j.jhazmat.2021.126217
[20] GAO Z, SHI L, YAN F, et al. Two-dimensional supramolecular polymers based on selectively recognized aromatic cation-π and donor-acceptor motifs for photocatalytic hydrogen evolution[J]. Angewandte Chemie International Edition, 2023, 62(21): e202302274. DOI: 10.1002/anie.202302274
[21] LI Y, WAN S, LIANG W, et al. D-A conjugated polymer/CdS S-scheme heterojunction with enhanced interfacial charge transfer for efficient photocatalytic hydrogen generation[J]. Small, 2024, 20(31): 2312104.
[22] ZHANG X L, YUAN N, LI Y, et al. Fabrication of new MIL-53(Fe)@TiO2 visible-light responsive adsorptive photocatalysts for efficient elimination of tetracycline[J]. Chemical Engineering Journal, 2022, 428: 131077. DOI: 10.1016/j.cej.2021.131077
-
期刊类型引用(1)
1. 章巧丽,王雅萍,孙万杰,易清风. 片状碳载金属纳米颗粒复合物的制备及其氧还原电活性(英文). 无机化学学报. 2023(02): 346-356 .  百度学术
百度学术
其他类型引用(3)
-
其他相关附件
-
目的
作为一种清洁、可持续的能源,氢能可以应对日益加剧的全球能源危机。目前,热化学转化、电解等技术被用于水分解制氢,但它们往往伴随着高能耗、复杂操作或高成本。太阳能驱动光催化水分解制氢是一种极具发展潜力的新途径,而受到广泛关注。寻找合适的半导体光催化剂一直是该领域的研究热点。CdZnS固溶体具有适宜的能带结构,能够为光催化水制氢提供合适的电位。然而,光吸收范围窄、载流子分离效率低、光生电荷迁移速率慢是现有CdZnS光催化材料存在的主要缺陷,严重限制了光催化活性的提高。构建异质结光催化剂可以有效地提高光催化效率。
方法采用溶剂热法制备了CdZnS固溶体、CeO/CdZnS异质结。利用XRD、SEM、XPS等表征手段对其样品的晶型、形貌、结构、元素组成等进行了表征。在可见光照射下,研究了CdZnS固溶体、CeO/CdZnS异质结光催化产氢性能。通EPR、DFT计算等研究了固溶体、异质结的内电场及光催化性能增强的机理。
结果XRD的衍射角偏移、衍射峰强度变化表明CdZnS复合材料不是简单的CdS和ZnS的混合物,而是一种固溶体相,CZS-0.3纳米颗粒与CeO形成复合材料。SEM和TEM分析表明CZS-0.3与CeO充分接触。XPS表征表明电子CeO的电子迁移到CdZnS。光催化结果表明CeO/CdZnS异质结具有优异的光催化产氢性能。电化学测试、UV-Vis DRS结果表明CeO/CdZnS异质结具有合适的氧化还原电位。DFT计算表明形成内电场,抑制了电子-空穴对的复合,加速了界面载流子的分离和转移。
结论(1)采用溶剂热法煅烧法制备了CdZnS固溶体和 y%CCZS-0.3异质结。(2)CdZnS固溶体和 y%CCZS-0.3异质结在可见光照射下能有效地实现析氢。10%CCZS-0.3异质结的析氢速率为7.89 mmol·g·h,分别是CeO、CdZnS固溶体的40.25、2.04倍。(3)CeO和CZS-0.3形成了Z型异质结结构,在10%CCZS-0.3异质结界面处形成内置电场,大大减少了光生载流子的重组,增加了光生电子的积累,从而提高了光催化活性。
-
作为一种清洁、可持续的能源,氢能可以应对日益加剧的全球能源危机。目前,热化学转化、电解等技术被用于水分解制氢,但它们往往伴随着高能耗、复杂操作或高成本。太阳能驱动光催化水分解制氢是一种极具发展潜力的新途径,而受到广泛关注。寻找合适的半导体光催化剂一直是该领域的研究热点。CdxZn1-xS固溶体具有适宜的能带结构,能够为光催化水制氢提供合适的电位。然而,光吸收范围窄、载流子分离效率低、光生电荷迁移速率慢是现有CdxZn1-xS光催化材料存在的主要缺陷,严重限制了光催化活性的提高。
本文采用溶剂热法制备了CdxZn1-xS固溶体、CeO2/CdxZn1-xS异质结,并利用XRD、SEM、XPS等表征手段对其样品的晶型、形貌、结构、元素组成等进行了表征。可见光照射下,研究了CdxZn1-xS固溶体、CeO2/CdxZn1-xS异质结产氢性能。Cd0.3Zn0.7S异质结的产氢速率为3.86 mmol·g-1·h-1,分别是CdS、ZnS的4.85、11.03倍。当CeO2负载比例为10%时,CeO2/Cd0.3Zn0.7S异质结具有最佳的光催化性能,产氢速率为7.89 mmol·g-1·h-1,分别是CeO2、Cd0.3Zn0.7S固溶体的40.25、2.04倍。光照下,CeO2的电子迁移到CdxZn1-xS,使得靠近CeO2的异质结界面部分带正电,而靠近CdxZn1-xS的异质结界面部分带负电,形成内电场,增强了载流子分离与迁移性能。
CeO2、y%CCZS-0.3的光催化产氢性能(a,b)
















计量
- 文章访问数: 232
- HTML全文浏览量: 119
- PDF下载量: 18
- 被引次数: 4
