

Molecular dynamics simulation of atomic migration and diffusion in composite interface for non-coherent metals
-
摘要:
深入探究不锈钢/碳钢金属界面原子扩散行为以及相变的发生发展规律,对于提升金属间冶金结合质量、实现产品性能调控具有重要意义。本文基于分子动力学材料计算方法,建立COMPASS力场下的不锈钢FCC-Fe和碳钢BCC-Fe晶胞模型;在热压缩高温保温和连续压缩两个阶段分别采用NVT和NPT系综,保温温度1423K,压应力分别为2GPa和4GPa;通过研究界面微观结构、均方位移分布、径向分布函数和界面元素分布模拟非共格金属界面结构演变行为。结果表明,在保温阶段,碳钢侧晶体发生BCC-Fe→FCC-Fe相变,空间群由P1向FM-3M的转变过程为无序长程扩散。在加载200ps弛豫结束时刻,不锈钢与碳钢侧原子相互嵌入,形成统一的面心立方晶体;且随着压力增加,界面结构以最密排的(111)晶面为单位产生大量的滑移和错排,两组元原子能够发生有效的扩散迁移。
Abstract:Pointing to the diffusion behavior of atom and the law of phase transition in metal interface for stainless steel and carbon steel, a further research is of great significance for improving the quality of metallurgical bonding and realizing the property regulation of product. In this paper, based on the kind of material calculation method of molecular dynamics, cell models including carbon steel BCC-Fe and stainless steel FCC-Fe were established apart under COMPASS force field. And NVT and NPT systems were also employed in the two different stages of high-temperature insulation with the temperature of 1423K and continuous compression with the stress of 2 GPa and 4 GPa, respectively. On this base, the change behavior of non-congruent metal interface was simulated through the indicators of interfacial microstructure, the mean-squared displacement distribution, the radial displacement function and the interface elemental distribution. The results show that the phase transition of BCC-Fe→FCC-Fe happens to occur in the carbon steel side during the high-temperature insulation stage, accompaning the space transition of P1→FM-3M in a disordered and long-range diffusion process. During the loading relaxation stage of 200ps, the boundary atoms are embedded in each other until the inferface forms a unified face-centered cubic crystal. With the increase of pressure, the interface structure produces a large number of slips and misalignments along the most densely-rowed crystal face of (111), and the two groups of atoms could success an effective diffusion and migration.
-
不锈钢/碳钢复合材料因其优异的力学性能和耐蚀性,广泛应用于核能核电、石油化工、海洋工程以及装甲制造等领域。与爆炸焊、堆焊等生产方法相比,热轧复合具有结合界面完整、生产效率高、安全环保等优势,是目前层状金属复合材料主要生产方法[1,2]。其复合过程为高压作用下金属剧烈塑性变形,同时还伴随着高温下界面原子键合扩散。各组分材料的金属流动协调变形行为以及原子扩散存在显著差异,严重影响金属复合材料的界面微观结构和力学性能[3,4]。因此,研究不锈钢/碳钢金属热轧复合机理和技术,不仅需要开展复合材料制备关键共性技术基础,揭示金属复合材料的变形行为、控制轧制与组织协同参数设计,还需要深入探究异质金属结合界面原子扩散行为以及相变的发生发展规律,这对于提升金属间冶金结合质量、实现产品性能调控具有重要意义。
目前,基于原子尺度模拟凝聚相界面变化是分子动力学(molecular dynamics,MD)方法的重要分支,涵盖了复合材料界面扩散反应、界面结构、界面力学性能以及界面失效等方面[5,6]。Degiacomi等[7]为评估界面分子动力学模拟的收敛性,通过模拟盒尺寸对双组分系统的影响发现,对于具有较小系统(或较小体积区域)仅仅需要大约几十纳秒就足以表征界面相互作用。Chen等[8]结合分子动力学模拟和经典扩散理论,分析Cu-Al在爆炸焊接过程中的原子扩散行为,发现原子扩散主要发生在焊接过程的卸载阶段;且扩散系数与碰撞速度正相关。王锐等[9]研究拉伸载荷下镀镍石墨烯/钛复合材料界面演化行为,结果表明由于镀层镍阻挡迫使复合界面位错密度提高,界面力学性能提高。Chen等[10]采用MD模拟技术,研究高温高压工况下Cu-Ag界面扩散过程,结果表明界面区域的厚度取决于应力大小,界面区域在扩散结合过程中变为非晶态组织。崔云峰等[11]基于分子动力学方法,对Cu/Al2Cu/Al体系界面扩散过程进行了模拟研究,发现界面过渡层厚度随保温温度的升高逐渐向两侧扩展。Luo等[12]通过构建Mo和Ti的MAEAM势,通过发现(111)晶面相较于(110)和(100)晶面更有利于原子扩散,揭示Mo-Ti界面的不对称扩散机制。Bhasker-Ranganath、Kumagai等[13,14]借助分子动力学方法,构建合理的界面模型与材料计算方法,分析温度、应力等工艺参数对面心立方、体心立方晶体位错迁移率的影响。虽然分子动力学在研究异质金属界面结构领域取得一定进展,但是由于异质金属复合界面微观结构变化复杂,迄今对于晶体结构变化和界面原子迁移扩散机理仍不明确,金属界面复合理论的协同作用尚不确定。因此,本文基于Materials Studio(MS)中的COMPASS力场下的分子动力学方法,构建不锈钢/碳钢复合界面模型,通过设计不同的应力工况,研究不同应力条件下的界面晶体结构变化和原子扩散迁移规律等,为复合产品工业制备和性能调控提供理论支撑。
1. 分子动力学模型构建
1.1 FCC-Fe和BCC-Fe晶胞模型构建
本文采用的覆层材料为304不锈钢,基体材料为Q235碳钢。为了搭建两种金属材料的界面模型,首先将两种结构的铁原子模型从MS内置材料库中导出,原始晶胞空间群及几何大小参数见表1。不锈钢层采用FCC-Fe作为基体晶胞,晶格参数为:a=b=c=
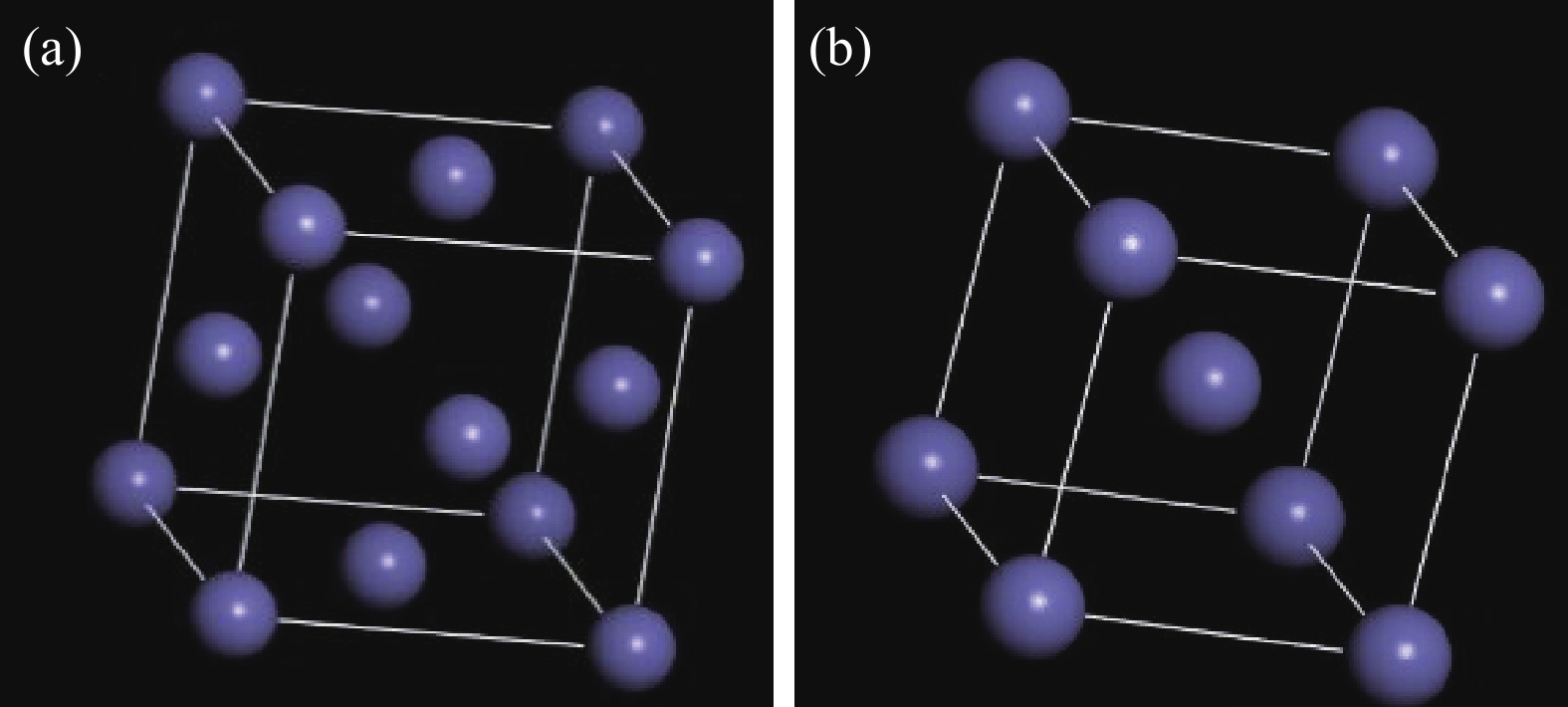
0.3582 nm、α=β=γ=90°;碳钢层采用BCC-Fe作为基体晶胞,晶格参数为:a=b=c=0.2859 nm、α=β=γ=90°[15]。由此建立了FM-3 M空间群结构的FCC-Fe原子晶胞、P1空间群结构的BCC-Fe原子晶胞和复合金属初始晶胞模型。三维立体模型如图1所示。表 1 基体晶胞的基本参数Table 1. Basic parameters of the unit cellAtom Group name Lattice constant/nm FCC-Fe FM-3 M a=b=c= 0.3582 BCC-Fe P1 a=b=c= 0.2859 ![]() 图 1 晶胞三维模型:(a) FCC-Fe;(b) BCC-FeFigure 1. Three-dimensional modeling of cell structures: (a) FCC-Fe; (b) BCC-Fe
图 1 晶胞三维模型:(a) FCC-Fe;(b) BCC-FeFigure 1. Three-dimensional modeling of cell structures: (a) FCC-Fe; (b) BCC-Fe构建不锈钢晶胞模型时,按照Fe∶Cr∶Ni=79∶19∶10的比例随机替代晶胞中的Fe原子;构建碳钢晶胞模型时,主要以Fe原子为主,C原子含量在0.2%以下,且处于固溶体点阵的间隙位置,对研究结果影响不大,建模时忽略C原子[16]。在本文的MD模拟中,晶胞由FCC层和BCC层组成。FCC层和BCC层的顶表面都在(100)平面上。晶胞中原子数的选择应使FCC-BCC界面的失配尽可能接近晶格常数的实验比值,并且结构尺寸是可控的。FCC层原子数选择12×12×4=576,BCC层原子数选择15×15×2=450,这是考虑到15/12=1.25,与FCC-Fe和BCC-Fe的晶格常数之比
1.2528 非常接近。FCC层与BCC层均为6层,则FCC层原子总数为3456 ,BCC层原子总数为2700 。周期性边界条件在两个横向(即x和y)方向[17]。按晶格失配率的计算标准来计算,这个界面模型的晶格失配为
OA方向的错配=0.3582×12−0.2859×150.3582×12=0.23% (1) OB方向的错配=0.3582×12−0.2859×150.3582×12=0.23% (2) 晶格失配率满足了界面模型的要求,因此可以成功创建成所需的界面[18]。
张清东等[19]在研究非共格界面对金属微观变形行为的影响时,构建的不同晶格结构的不锈钢/碳钢晶胞模型包含
16384 个原子,而王宝峰等[20]构建的铜/铝热轧复合界面模型包含5300 个原子。因此,为了保障压力传导的均匀性,盒子尺寸不宜过大。本文所构建的模型实际尺寸为4.2937 nm×4.2937 nm×4.1258 nm,共6156 个原子。FeCrNi/Fe模型的初始构型如图2所示。![]() 图 2 FeCrNi/Fe模型的初始构型:(a)左视图;(b)主视图Figure 2. Initial configuration of FeCrNi/Fe: (a) Left view; (b) Front view
图 2 FeCrNi/Fe模型的初始构型:(a)左视图;(b)主视图Figure 2. Initial configuration of FeCrNi/Fe: (a) Left view; (b) Front view1.2 边界条件设定
COMPASS力场是适合凝聚态应用的一个全新的分子力场,广泛覆盖共价分子和使用各种非共价模型的无机材料,包括最常见的有机物、聚合物、金属、金属离子和金属氧化物等,能够准确预测分离相和凝聚相分子的分子结构、振动频率、构象能、偶极矩、晶体结构、状态方程和内聚能密度[21],同时综合考虑范德华力以及库仑力等。基于它的优势和特性,殷开梁等[22]关于氢键问题,王泉等[23]关于铁原子与聚合物/碳纳米管复合材料的摩擦问题,以及肖继军等[24]关于TATB基PBX结合能和力学性能研究中都采用了COMPASS力场。
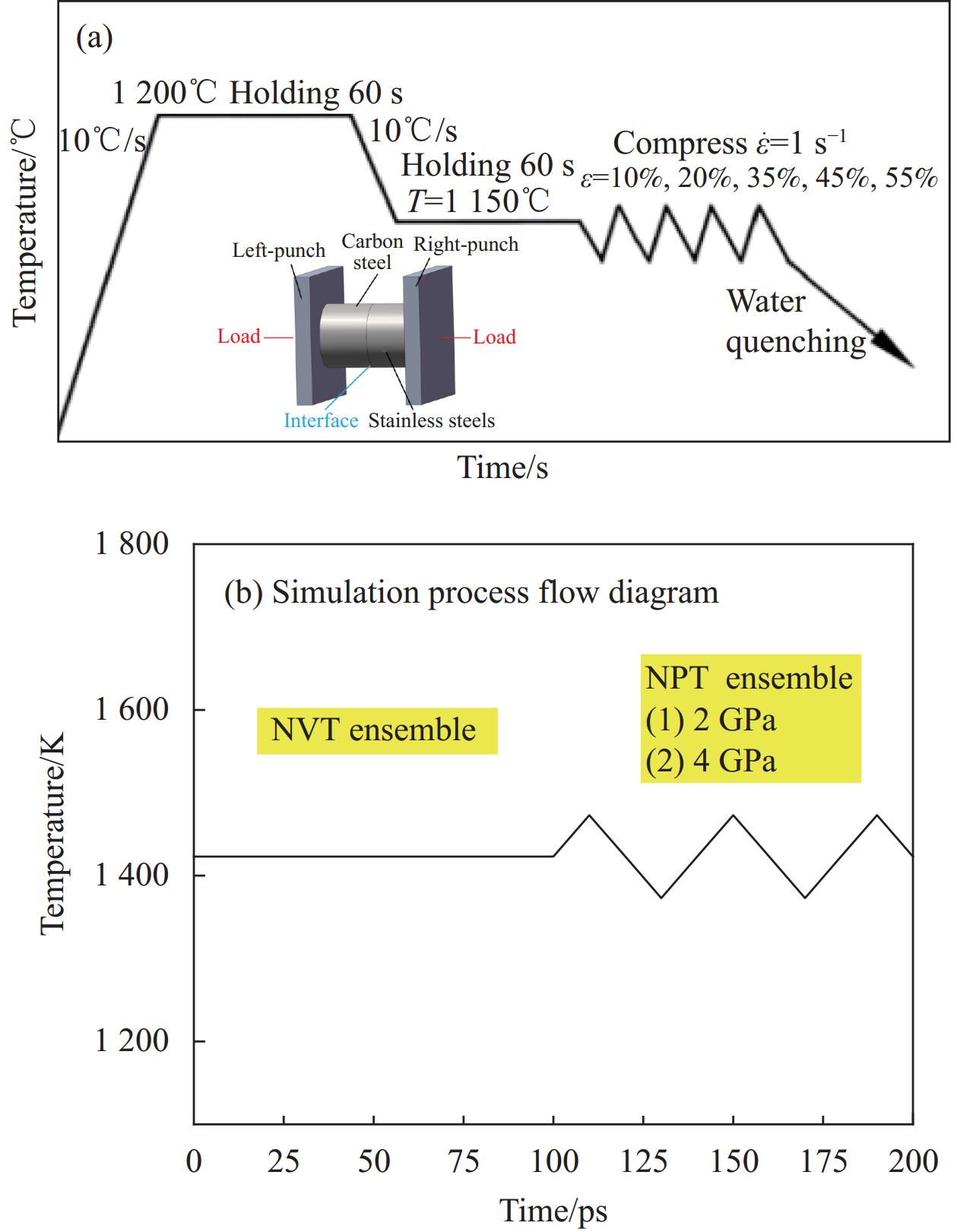
在系综的选择与设定方面,采取NVT(等温等体积)和NPT(等温等压)系综。它们是由Anderson通过对拉格朗日方程的重新推导,实现了恒温和恒压体系在分子动力学模拟的广泛应用。热压缩实验可以看作高温保温(恒温)和连续压缩(恒压)两个阶段,如图3(a)所示。因此,在保温阶段,采用NVT系综工况,体系的原子数量n、体积v和温度t保持不变,且模拟盒子box的几何尺寸不会发生变化;将保温温度设定为
1423 K,这是由于一般来说,扩散结合所需温度基本在0.6~0.8 Tm(其中Tm表示相关材料的熔点)之间,高温对界面扩散具有重要的促进作用。![]() 图 3 高温热压缩工艺及系综示意图:(a)高温热压缩工艺;(b)NVT和NPT系综Figure 3. Schematic diagram of thermal compression process and NVT and NPT systems: (a) Thermal compression process; (b) NVT and NPT systems
图 3 高温热压缩工艺及系综示意图:(a)高温热压缩工艺;(b)NVT和NPT系综Figure 3. Schematic diagram of thermal compression process and NVT and NPT systems: (a) Thermal compression process; (b) NVT and NPT systems在连续压缩阶段,采用NPT(等温等压)系综工况,保证体系的原子数量n、压强p和温度t保持不变,不同的是,NPT系综下box的尺寸可以发生变化。采用Souza-Martins控压方法,设计OC方向的单向受压工况,压应力分别为2 GPa、4 GPa。时间步长设定为1 fs,总模拟时间设定为200 ps,如图3(b)所示。
2. 模拟结果分析
2.1 复合界面结构演变
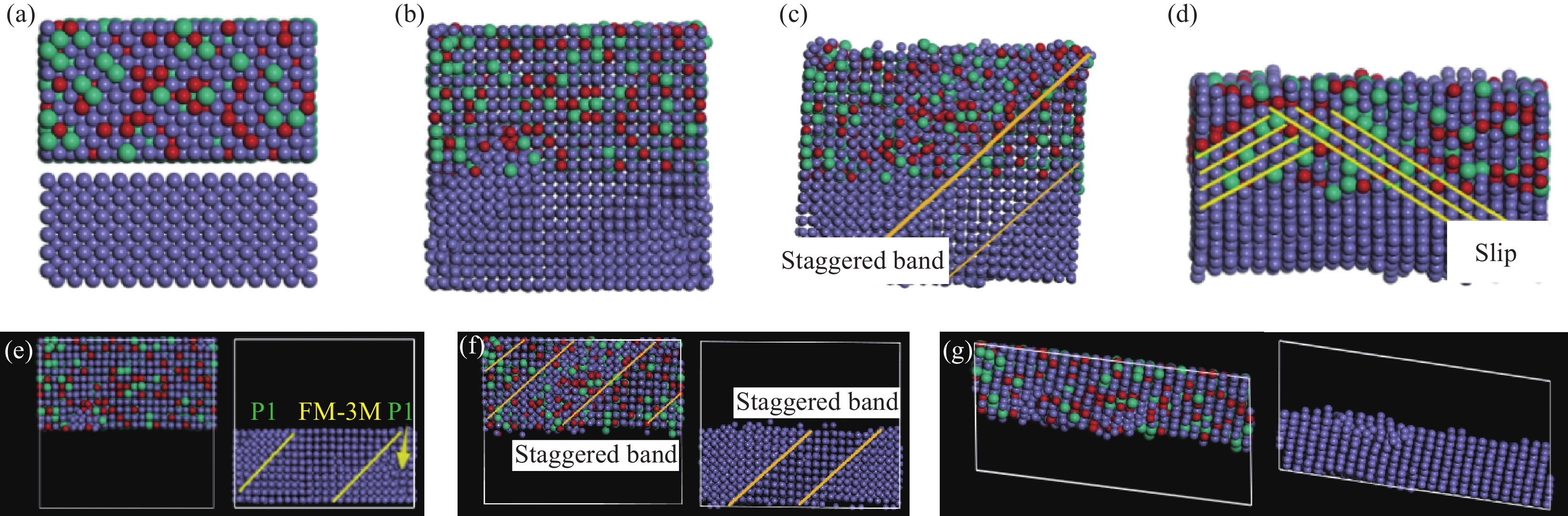
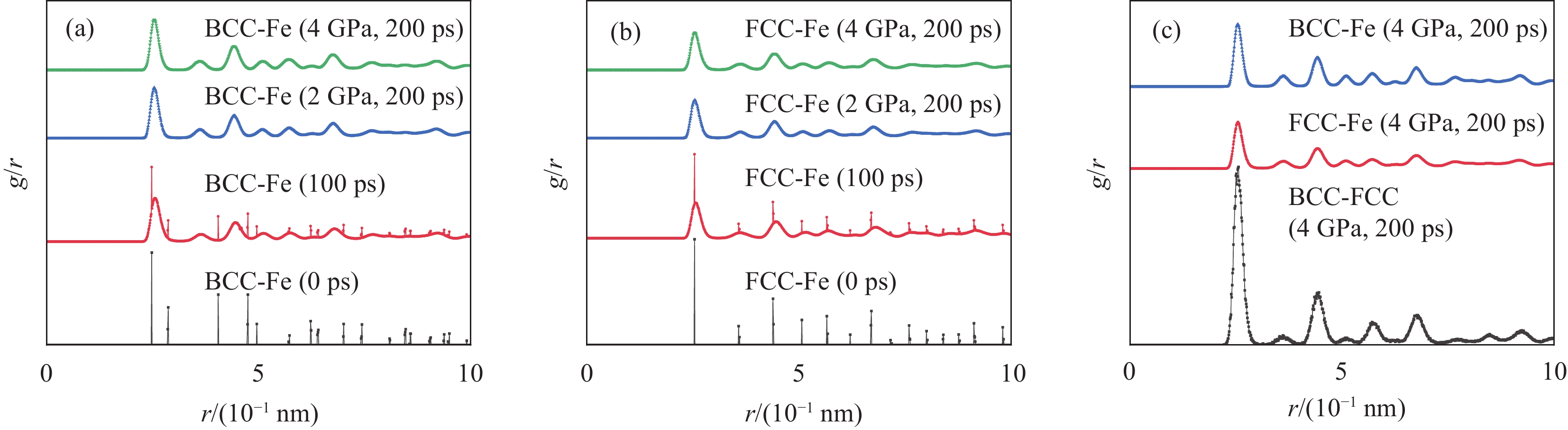
如图所示为界面结构模型。在0 ps初始阶段,如图4(a)所示,不锈钢侧和碳钢侧的原子被真空层分离。在100 ps(对应NVT系综)驰豫结束时刻,如图4(b)、4(e)所示,可以看出受高温影响,碳钢侧BCC-Fe晶体发生相变,开始向FCC-Fe晶体转变,空间群由P1向FM-3 M的转变过程为无序长程扩散。
![]() 图 4 不同阶段304/Q235界面结构模型:(a) 0 ps;(b) 100 ps;(c) 2 GPa, 200 ps;(d) 4 GPa, 200 ps;(e) 100 ps, 两侧;(f) 2 GPa, 200 ps, 两侧;(g) 4 GPa, 200 ps, 两侧Figure 4. Models of 304/Q235 interface structure in different stages (a) 0 ps (b) 100 ps (c) 2 GPa, 200 ps (d) 4 GPa, 200 ps (e) 100 ps, two sdes (f) 2 GPa, 200 ps, two sdes (g) 4 GPa, 200 ps, two sdes
图 4 不同阶段304/Q235界面结构模型:(a) 0 ps;(b) 100 ps;(c) 2 GPa, 200 ps;(d) 4 GPa, 200 ps;(e) 100 ps, 两侧;(f) 2 GPa, 200 ps, 两侧;(g) 4 GPa, 200 ps, 两侧Figure 4. Models of 304/Q235 interface structure in different stages (a) 0 ps (b) 100 ps (c) 2 GPa, 200 ps (d) 4 GPa, 200 ps (e) 100 ps, two sdes (f) 2 GPa, 200 ps, two sdes (g) 4 GPa, 200 ps, two sdes探究金属键能与熔点的关系发现[25,26],熔点越高,金属键越强。不锈钢熔点约
1375 ~1530 ℃,碳钢熔点约1210 ~1370 ℃,可见不锈钢侧原子间的键合能力强于碳钢原子间的键合能力。因此碳钢侧晶体结构在升温过程中不稳定,原子热振动剧烈,从而为原子迁移扩散提供扩散通道。随着碳钢侧相变的进行,不锈钢侧晶界原子紊乱,存在明显的错排现象。然而不锈钢侧FCC-Fe晶体受到Cr、Ni等合金元素的固溶强化
作用,晶体结构并没有发生明显变形,此时碳钢与不锈钢之间存在着明显的晶界。李硕[27]等人在研究不锈钢/碳钢层合板轧制复合机理中也提到对于非共格界面,由于晶格常数的失配以及不同金属晶格结构存在差异,界面对位错起到阻碍和吸收作用,使得位错在变形过程中不能穿过界面。按照热压缩仿真流程,在高温压缩阶段,采用NPT系综,
1423 K高温下,对晶胞分别施加OC向2 GPa、4 GPa的压应力。在200 ps弛豫结束时刻,如图4(c)、4(f)所示为,在2 GPa压应力作用下,晶胞尺寸由4.1258 nm被压缩至3.7366 nm(应变约为0.1),此时不锈钢侧与碳钢侧原子相互嵌入,无明显界面区分,形成了统一的面心立方晶体。在这个过程中,随着BCC-Fe晶体相变产生的应力水平升高,内能增加,不锈钢侧的面心立方固溶体组成的超级胞的滑移系也开始启动,不锈钢侧面心立方固溶体参与到变形之中,晶界之间的应力得到释放且统一后的晶体中沿着(111)晶面出现了错排带(如图所示)但错排带原子还未有足够能量越过势垒形成滑移。这与张清东[19]等人研究非共格界面对金属微观变形行为的影响得出的结论吻合,即双金属界面为非共格界面模式的情况下,在弛豫结束后碳钢基体(模型下半部分)内有大量位错线产生,不锈钢基体靠近界面处开始有位错出现,这意味着不锈钢基体开始塑性变形。当压应力升至4 GPa时,如图4(d)、4(g)所示,界面结构产生显著的塑性变形,组元尺寸由
4.1258 nm被压缩至2.5289 nm(应变约0.4)。在巨大压应力作用下,界面结构以最密排的(111)晶面为单位产生了大量的滑移,同时伴有错排,广泛的滑移是塑性变形的主要来源,两组元原子通过滑移错排以及借助滑移产生的空位缺陷进行了有效的扩散迁移,出现了较为良好且均匀的复合界面结构,此时碳钢与不锈钢之间难以分辨界限,已经产生了良好的冶金结合界面。2.2 均方位移分布
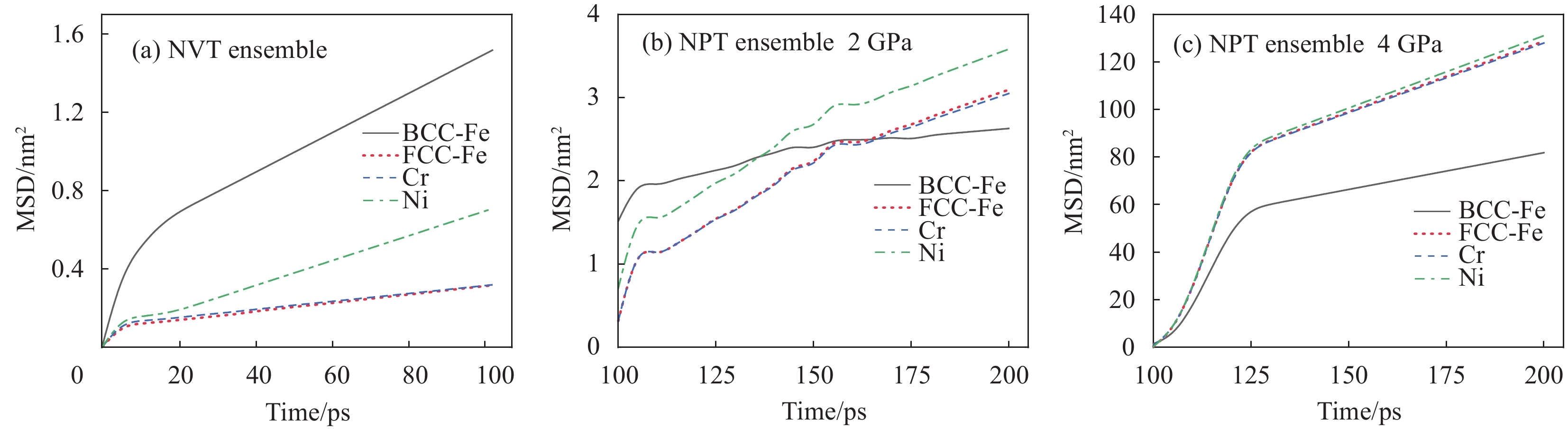
均方位移(Mean Square Displacement,MSD)不仅是用于描述微观粒子热运动的重要物理量,还可以用于描述宏观物体的振动现象。它是指物体在振动过程中,微观粒子离开平衡位置的平均距离的平方。图5为不同阶段不锈钢侧和碳钢侧原子的均方位移曲线。在NVT阶段,如图5(a)所示,不锈钢侧原子Fe、Cr、Ni的MSD约为0.3 nm2、0.3 nm2、0.8 nm2,碳钢侧Fe原子的MSD曲线虽然呈指数级上升,但最大值也仅约为1.5 nm2。表明此阶段原子不发生扩散,只是在平衡位置振动。
![]() 图 5 304/Q235界面各原子均方位移曲线图:(a)NVT系综;(b)NPT系综, 2 GPa;(c) NPT系综, 4 GPaFigure 5. MSD of different atoms in 304/Q235 inteface: (a)NVT ensemble; (b)NPT system, 2 GPa; (c)NPT system, 4 GPa
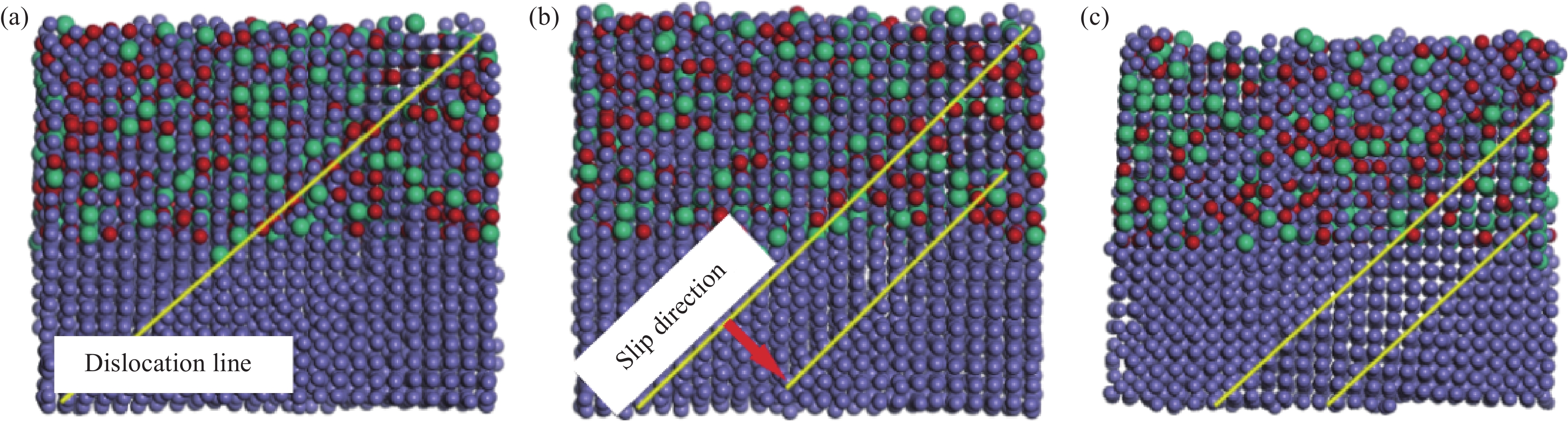
图 5 304/Q235界面各原子均方位移曲线图:(a)NVT系综;(b)NPT系综, 2 GPa;(c) NPT系综, 4 GPaFigure 5. MSD of different atoms in 304/Q235 inteface: (a)NVT ensemble; (b)NPT system, 2 GPa; (c)NPT system, 4 GPa在NPT加载阶段,2 GPa的单向压应力工况下,如图5(b)所示,在100~175 ps之间,碳钢侧的MSD始终高于不锈钢侧,这是由于碳钢侧空间群的转变持续进行,此时Fe原子的振幅显著;塑性变形主要发生在碳钢侧,如图6(a)、6(b)所示;然而在加载过程中,碳钢侧原子的迁移逐渐受到不锈钢侧FCC-Fe固溶体晶界的阻挡,引起位错的塞积。界面-位错机制指明晶界将对位错发展起到阻挡、弯曲甚至消失等抑制作用[28,29],因此界面会严重阻碍了位错在变形过程中的发展,致使MSD升高变得缓慢。
![]() 图 6 NPT系综2 GPa下的304/Q235界面结构演变:(a)150 ps;(b)170 ps;(c)190 psFigure 6. Interfacial structural evolution of 304/Q235 at 2 GPa in NPT system: (a)150 ps; (b)170 ps; (c)190 ps
图 6 NPT系综2 GPa下的304/Q235界面结构演变:(a)150 ps;(b)170 ps;(c)190 psFigure 6. Interfacial structural evolution of 304/Q235 at 2 GPa in NPT system: (a)150 ps; (b)170 ps; (c)190 ps当弛豫到175~200 ps时,此时碳钢侧完成空间群的转变,不锈钢侧开始出现位错,位错的出现使得原子间的运动更加容易。随着弛豫过程的进行,晶胞内的位错线由(111)晶面生成,同时在垂直于此晶面的方向缓慢向下滑移,如图6(c)所示。在NPT加载阶段,4 GPa的单向压力作用下,如图5(c)所示,界面两侧MSD数值攀高明显,这是由于不锈钢和碳钢组元界面已经开始出现连续滑移倾向,错排带原子有足够能量越过势垒形成滑移。此外,相比于碳钢侧,不锈钢侧各原子半径不同,原子间键能大小不同,在高压作用下原子热振动更为剧烈。
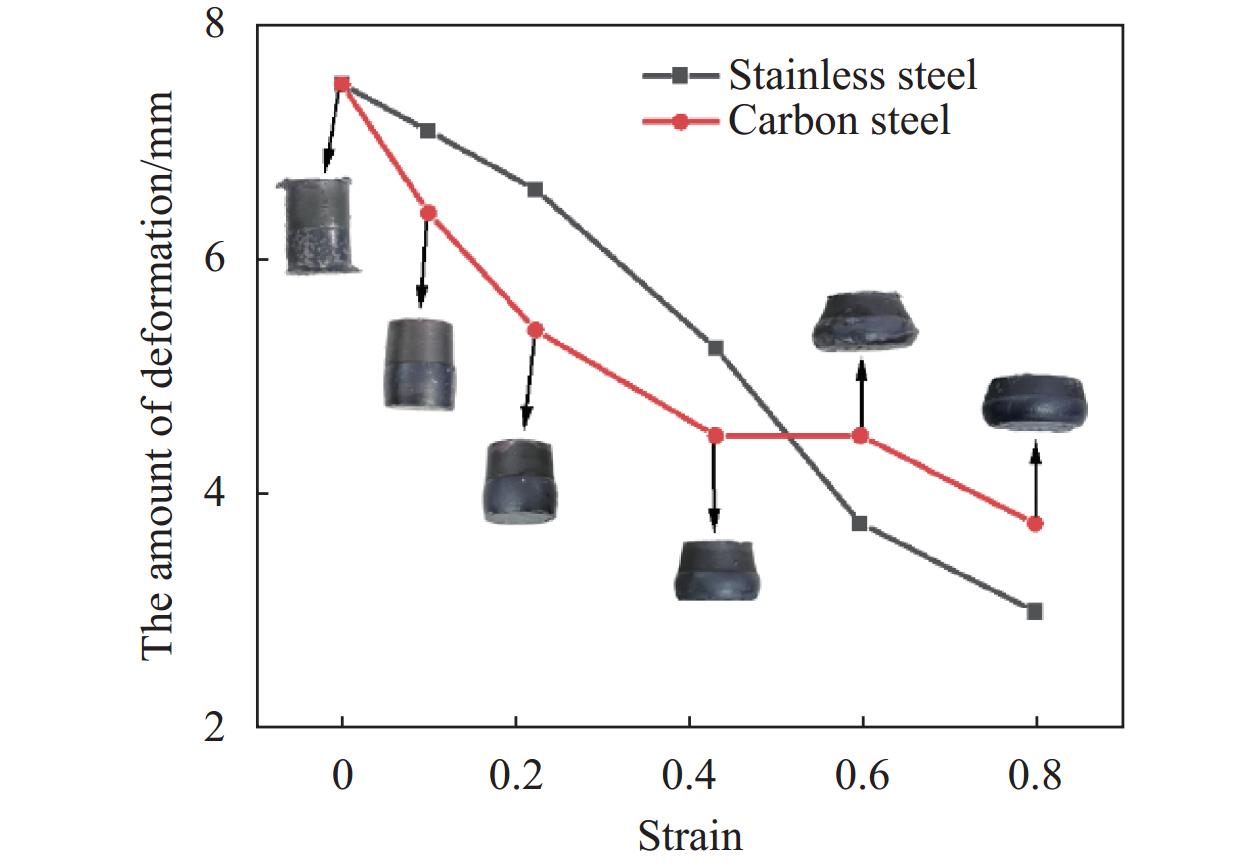
为了进一步探究双金属在不同压力、不同应变工况下的塑性变形行为差异,本文采用Gleeble-3800热力模拟机开展304不锈钢/Q235碳钢复合金属高温压缩实验。坯料高度均为7.5 mm,直径均为10 mm。为了保证良好的冶金结合效果,在组坯之前,采用砂纸将Q235碳钢和304不锈钢表面打磨,保证待复合表面以及上下端面的平整度和光洁度,露出内部新鲜金属;其次用无水乙醇洗去表面油污,保证表面清洁。变形温度设为
1150 ℃,应变率1 s−1,真应变0.1~0.8,热压缩过程中不锈钢/碳钢两侧基体厚度变化如图7所示。可以看出,当真应变较小时(0.1~0.5),由于高温下碳钢变形抗力低,碳钢变形量高于不锈钢;随着真应变增大,碳钢内部位错密度增加,且位错迁移受到不锈钢阻碍,因而变形受到限制;此时不锈钢侧滑移系开始启动,其变形能力逐渐增大。当总应变约为0.5时,两侧变形程度变化基本相当。随着真应变增大(>0.5),不锈钢的变形程度高于碳钢。因此,随着应变的不断增大,不锈钢和碳钢的厚度呈现出交替变化的趋势。从上面分子动力学构建的模型仿真结果来看,在2 GPa作用力下,碳钢BCC晶胞和不锈钢FCC晶胞变形量分别为8.9%、6.7%;在4 GPa作用力下,分别为32.7%、33.7%。可见仿真结果与热压缩复合厚度变化规律是吻合的。因此可以说明界面微观结构演变和位错变化在一定程度上能够诱发异质金属塑性变形差异。此外,模拟和试验数据难以完全拟合的原因是由于分子动力学理想化假设金属材料晶体无缺陷模型,这将不可避免的造成模拟偏差,但也是该方法目前难以克服的不足。![]() 图 7 热压缩实验中各基材厚度的变化Figure 7. Thickness variation of each metal during thermal compression experiments
图 7 热压缩实验中各基材厚度的变化Figure 7. Thickness variation of each metal during thermal compression experiments2.3 径向分布函数
径向分布函数(Radial Distribution Function, RDF)可以分析各粒子之间的作用关系和微观分布情况,代表给定粒子的位置后其它粒子在空间的分布概率,是反映材料微观结构的特征物理量,其强度可以用来表示原子排布的有序性,在本模拟中可以反映界面组元的结合程度。如图8(a)、8(b)所示,在0 ps时,两侧Fe原子的径向分布函数曲线呈高而窄的形状,表明加载前,碳钢和不锈钢两侧具有规则的晶体结构[30]。
![]() 图 8 不同阶段径向分布函数:(a)碳钢侧;(b)不锈钢侧;(c)结合界面Figure 8. Radial distribution function in different stages: (a) Side of carbon steel; (b) Side of stainless steel; (c) Interface
图 8 不同阶段径向分布函数:(a)碳钢侧;(b)不锈钢侧;(c)结合界面Figure 8. Radial distribution function in different stages: (a) Side of carbon steel; (b) Side of stainless steel; (c) Interface如图8(a)所示,在100 ps时,碳钢侧Fe原子的RDF峰值位置与0 ps时部分重合,同时也产生了新的峰值,说明此时碳钢侧部分晶体完成了空间群由P1向FM-3 M的转变[31]。加载完成到达200 ps时,2 GPa和4 GPa压应力作用下,碳钢侧Fe原子的径向分布函数曲线变短变宽,表明Fe原子在高温高压下不再是完整的规则晶体结构[32]。这主要是由于碳钢侧晶胞经受塑性变形,原子的不规则运动加剧,离开了原来的平衡位置,导致结构的无序移位。
然而,在两种工况的不同弛豫阶段,不锈钢侧Fe原子的RDF峰值位置与0 ps基本重合,如图8(b)所示,表明整个热压缩过程中,不锈钢侧无晶体结构的转变。由图8(c)界面层的径向分布函数可以看出,经过加载,界面结合层的RDF值的所有特征峰均高于两侧的特征峰值,表明界面原子的结构统一性高于两侧任一方晶胞,此时界面结构统一性最高。
2.4 界面元素扩散行为
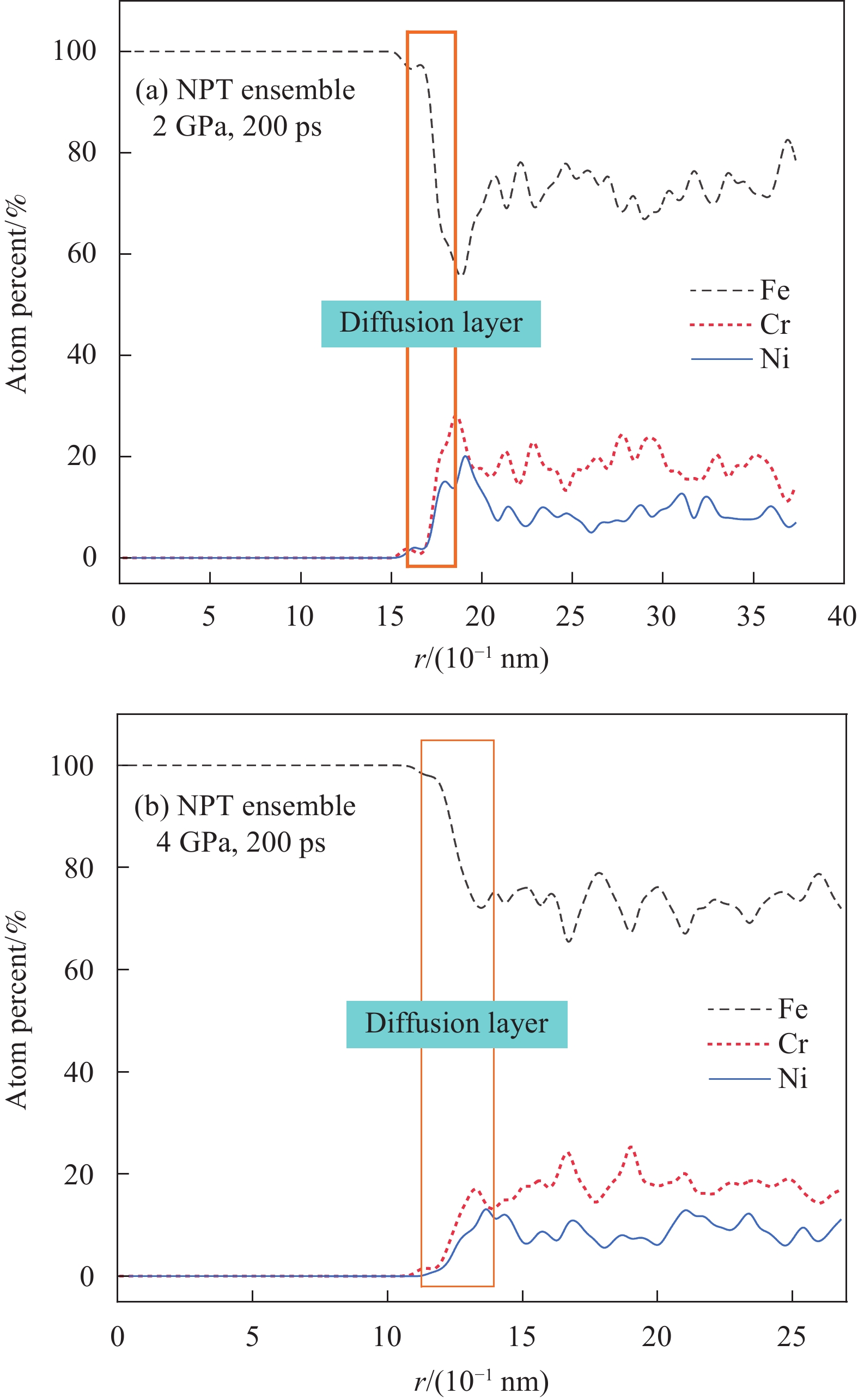
为了进一步定量分析不锈钢/碳钢界面原子扩散层厚度以及界面沿(001)晶界方向Fe、Cr、Ni原子的相对含量。扩散区定义为界面附近原子相对含量超过5%的区域[33,34]。如图9(a)、9(b)所示,在施加2 GPa、4 GPa压力工况下原子的扩散层厚度分别为
2.8173 ×10−1 nm和4.3787 ×10−1 nm。可见随着施加压力的增大,扩散层的厚度显著增加。需要说明的是,碳钢与不锈钢之间只有少数原子产生扩散迁移,原子的扩散迁移主要基于界面间的滑移产生,这是因为构建的界面为理想模型,没有空位、位错等晶体缺陷,而Fe、Cr、Ni原子半径相近,难以产生间隙扩散,且碳钢与不锈钢两组元在该温度下晶态保持良好,并未产生熔融,造成界面原子扩散距离近、扩散数目少[35]。![]() 图 9 304/Q235界面元素浓度分布仿真曲线:(a) NPT系综, 2 GPa, 200 ps;(b) NPT系综,4 GPa, 200 psFigure 9. Simulation curves of elemental concentration distribution in 304/Q235 interface: (a) NPT system, 2 GPa, 200 ps; (b) NPT system, 4 GPa, 200 ps
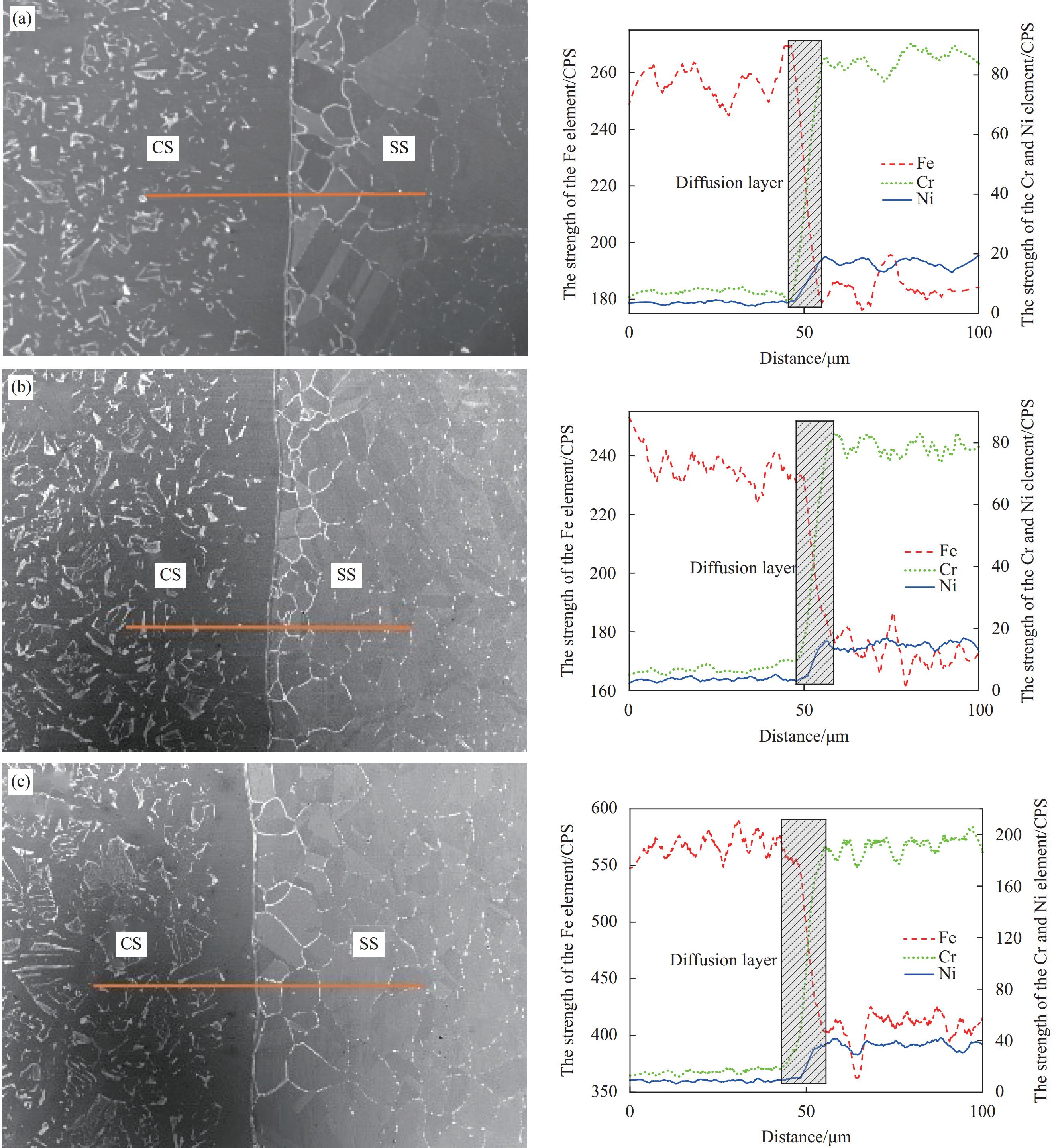
图 9 304/Q235界面元素浓度分布仿真曲线:(a) NPT系综, 2 GPa, 200 ps;(b) NPT系综,4 GPa, 200 psFigure 9. Simulation curves of elemental concentration distribution in 304/Q235 interface: (a) NPT system, 2 GPa, 200 ps; (b) NPT system, 4 GPa, 200 ps图10为不锈钢/碳钢复合金属高温压缩界面元素扩散实验结果,压下量为20%、35%和55%时,形成宽度约为6.32、8.57和10.25μm的扩散层。可以看出随着压下率增大,复合界面宽度增加,且元素浓度变化趋于平缓且曲线更为平滑。
![]() 图 10 304/Q235界面元素扩散分布实验结果:(a) 20%;(b) 35%;(c) 55%Figure 10. Experiment results of elemental diffusion in 304/Q235 interface: (a) 20%; (b) 35%; (c) 55%
图 10 304/Q235界面元素扩散分布实验结果:(a) 20%;(b) 35%;(c) 55%Figure 10. Experiment results of elemental diffusion in 304/Q235 interface: (a) 20%; (b) 35%; (c) 55%图11为
1150 ℃时不同压下率对元素扩散距离的影响。随着压下率增大,变形程度加剧,引起缺陷诱导效应和再结晶程度增加,晶粒细化,晶界数量增加,扩散通道增多,有利于元素扩散[36];同时,双金属界面结合致密紧实,使得元素扩散路径缩短。压下率达到55%时,界面平滑,无微裂纹、空洞等缺陷,形成良好的界面结合质量。![]() 图 11
图 111150 ℃下不同变形量对304/Q235界面元素扩散距离的影响Figure 11. Effect of different deformation degree on the diffusion distance of 304/Q235 interface elements at1150 ℃综上,本文在分子动力学模拟过程中,将总模拟时间设为200 ps。其中,在0~100 ps对应热压缩恒温保温阶段;在100~200 ps对应热压缩加载阶段,施加的压应力分别为2 GP和4 GP。在NPT加载初期,即在100~175 ps区间,碳钢侧空间群转变持续进行,塑性变形主要发生在碳钢侧。到175~200 ps区间,此时碳钢侧完成空间群转变,不锈钢侧开始出现位错。这个过程符合N.Bay理论指出的外界压力使两种金属的接触面产生不协调变形。
此外,径向分布函数显示,弛豫后期,在高温高压作用下碳钢和不锈钢不再是完整的规则晶体结构,界面结合层RDF值的所有特征峰均高于两侧特征峰值。这主要归功于界面不断地互相接近,直到原子级距离,在界面产生协调变形的结果。界面元素扩散模拟显示,在压应力作用下,原子间运动更为剧烈,界面具有明显的扩散层。这个过程符合N.Bay理论指出的压力使两种金属的接触面接近到原子级距离,并由原子吸附产生大量结合点,最终实现异种金属元素扩散,实现冶金结合。
3. 结 论
利用分子动力学模拟软件Materials Studio,建立了COMPASS力场下的不锈钢/碳钢界面模型,并通过加载不同的压应力,分析界面微观结构演变和原子迁移扩散机制,得到以下结论:
(1)在温度
1423 K工况,在0 ps初始阶段,不锈钢和碳钢侧的原子被真空层分离。在100 ps驰豫结束时刻,受高温影响,碳钢侧晶体发生BCC-Fe→FCC-Fe转变,空间群由P1向FM-3 M的转变过程为无序长程扩散;然而不锈钢侧FCC-Fe晶体受到Cr、Ni等合金元素的固溶强化作用,晶体结构没有发生明显变形,此时界面之间存在明显的晶界;(2)在温度
1423 K工况,沿轴向对复合模型施加2 GPa、4 GPa的压应力,随着BCC-Fe晶体相变产生的内能增加,不锈钢侧面心立方固溶体组成的超级胞滑移系开始启动,不锈钢参与到变形中;且在大压应力(4 GPa)作用下,界面结构以最密排的(111)晶面为单位产生了大量的滑移和错排,两组元原子能够进行有效的扩散迁移。分子动力学模拟异质金属的复合过程符合学术界普遍认可的N.Bay理论;(3)宏观试验结果表明,不同工况下复合材料的宏观变形和微观元素扩散规律与分子动力学微观结果演变规律完全一致,因此文中所采用的Materials Studio中的COMPASS力场和NVT、NPT系综是完全适用于模拟非共格异质金属界面微观结构变化和元素扩散迁移行为等的变化规律,对于揭示异质金属复合机理具有重要意义。
-
![]()
图 1 晶胞三维模型:(a) FCC-Fe;(b) BCC-Fe
Figure 1. Three-dimensional modeling of cell structures: (a) FCC-Fe; (b) BCC-Fe
![]()
图 2 FeCrNi/Fe模型的初始构型:(a)左视图;(b)主视图
Figure 2. Initial configuration of FeCrNi/Fe: (a) Left view; (b) Front view
![]()
图 3 高温热压缩工艺及系综示意图:(a)高温热压缩工艺;(b)NVT和NPT系综
Figure 3. Schematic diagram of thermal compression process and NVT and NPT systems: (a) Thermal compression process; (b) NVT and NPT systems
![]()
图 4 不同阶段304/Q235界面结构模型:(a) 0 ps;(b) 100 ps;(c) 2 GPa, 200 ps;(d) 4 GPa, 200 ps;(e) 100 ps, 两侧;(f) 2 GPa, 200 ps, 两侧;(g) 4 GPa, 200 ps, 两侧
Figure 4. Models of 304/Q235 interface structure in different stages (a) 0 ps (b) 100 ps (c) 2 GPa, 200 ps (d) 4 GPa, 200 ps (e) 100 ps, two sdes (f) 2 GPa, 200 ps, two sdes (g) 4 GPa, 200 ps, two sdes
![]()
图 5 304/Q235界面各原子均方位移曲线图:(a)NVT系综;(b)NPT系综, 2 GPa;(c) NPT系综, 4 GPa
Figure 5. MSD of different atoms in 304/Q235 inteface: (a)NVT ensemble; (b)NPT system, 2 GPa; (c)NPT system, 4 GPa
![]()
图 6 NPT系综2 GPa下的304/Q235界面结构演变:(a)150 ps;(b)170 ps;(c)190 ps
Figure 6. Interfacial structural evolution of 304/Q235 at 2 GPa in NPT system: (a)150 ps; (b)170 ps; (c)190 ps
![]()
图 7 热压缩实验中各基材厚度的变化
Figure 7. Thickness variation of each metal during thermal compression experiments
![]()
图 8 不同阶段径向分布函数:(a)碳钢侧;(b)不锈钢侧;(c)结合界面
Figure 8. Radial distribution function in different stages: (a) Side of carbon steel; (b) Side of stainless steel; (c) Interface
![]()
图 9 304/Q235界面元素浓度分布仿真曲线:(a) NPT系综, 2 GPa, 200 ps;(b) NPT系综,4 GPa, 200 ps
Figure 9. Simulation curves of elemental concentration distribution in 304/Q235 interface: (a) NPT system, 2 GPa, 200 ps; (b) NPT system, 4 GPa, 200 ps
![]()
图 10 304/Q235界面元素扩散分布实验结果:(a) 20%;(b) 35%;(c) 55%
Figure 10. Experiment results of elemental diffusion in 304/Q235 interface: (a) 20%; (b) 35%; (c) 55%
![]()
图 11
1150 ℃下不同变形量对304/Q235界面元素扩散距离的影响Figure 11. Effect of different deformation degree on the diffusion distance of 304/Q235 interface elements at
1150 ℃表 1 基体晶胞的基本参数
Table 1 Basic parameters of the unit cell
Atom Group name Lattice constant/nm FCC-Fe FM-3 M a=b=c= 0.3582 BCC-Fe P1 a=b=c= 0.2859  下载: 导出CSV
下载: 导出CSV
-
[1] Jiang J, Ding H, Luo Z, et al. Interfacial microstructure and mechanical properties of stainless steel clad plate prepared by vacuum hot rolling[J]. Journal of Iron and Steel Research International, 2018, 25: 732-738. DOI: 10.1007/s42243-018-0090-7
[2] Jin J C, Cho S, Kim K, et al. Microstructures and intergranular corrosion resistances of hot-rolled austenitic stainless steel clad plates[J]. Journal of Materials Research and Technology, 2023, 26: 1-13. DOI: 10.1016/j.jmrt.2023.07.192
[3] 班慧勇, 梅镱潇, 石永久. 不锈钢复合钢材钢结构研究进展[J]. 工程力学, 2021, 38(6): 1-23. DOI: 10.6052/j.issn.1000-4750.2020.04.ST01 BAN Huiyong, MEI Yixiao, SHI Yongjiu. Research progress of stainless steel compo-site steel structure[J]. Engineering Mechan-ics, 2021, 38(6): 1-23(in Chinese). DOI: 10.6052/j.issn.1000-4750.2020.04.ST01
[4] Mudhaffar M A, Saleh N A, Aassy A. Influence of hot clad rolling process parameters on life cycle of reinforced bar of stainless steel carbon steel bars[J]. Procedia Manufacturing, 2017, 8: 353-360. DOI: 10.1016/j.promfg.2017.02.045
[5] 潘帅航, 尹念, 张执南. 微动界面连续干摩擦行为的分子动力学模拟[J]. 机械工程学报, 2018, 54(3): 82-87. DOI: 10.3901/JME.2018.03.082 PAN Shuaihang, YIN Nian, ZHANG Zhinan. Molecular dynamics simulation of continu-ous dry friction behavior of micromotor in-terfaces[J]. Journal of Mechanical Engineer-ing, 2018, 54(3): 82-87(in Chinese). DOI: 10.3901/JME.2018.03.082
[6] 王路生. 基于分子动力学与有限元方法的金属材料变形及失效的多尺度模拟[D]. 重庆: 重庆理工大学, 2018. WANG Lusheng. Multi-scale simulation of deformation and failure of metal materials based on molecular dynamics and finite element method [D]. Chongqing: Chongqing University of Technology, 2018. (in Chinese)
[7] Degiacomi MT, Tian S, Greenwell HC, et al. DynDen: Assessing convergence of molecular dynamics simulations of interfaces[J]. Computer Physics Communications, 2021, 269: 108126. DOI: 10.1016/j.cpc.2021.108126
[8] Chen S Y, Wu Z W, Liu K X, et al. Atomic diffusion behavior in Cu-Al explosive welding process[J]. Journal of Applied physics, 2013, 113(4): 044901-044906. DOI: 10.1063/1.4775788
[9] 王锐, 韩秀丽, 康鹏超, 等. 镀镍石墨烯/钛复合材料界面力学行为的分子动力学研究[J]. 精密成形工程, 2024, 16(4): 45-52. DOI: 10.3969/j.issn.1674-6457.2024.04.006 WANG Rui, HAN Xiuli, KANG Pengchao, et al. Molecular Dynamics study of interfacial Mechanical Behavior of Nickel-coated graphene/titanium Composites[J]. Precision Forming Engineering, 2024, 16(4): 45-52(in Chinese). DOI: 10.3969/j.issn.1674-6457.2024.04.006
[10] Chen S D, Soh A K, Ke F J. Molecular dynamics modeling of diffusion bonding[J]. Scripta Materialia, 2005, 52(11): 1135-1140. DOI: 10.1016/j.scriptamat.2005.02.004
[11] 崔云峰, 王文焱, 谢敬佩, 等. 分子动力学分析Cu/Al2Cu/Al体系的扩散过程[J]. 材料热处理学报, 2023,44(11): 167−175. DOI: 10.13289/j.issn.1009-6264.2023-0136 CUI Yunfeng, WANG Wenyan, XIE Jingpei, et al. Cu/Al2Cu/Al system for molecular dynamics analysis of diffusion process[J]. Journal of materials, heat treatment,2023,44(11): 167−175. DOI: 10.13289/j.issn.1009-6264.2023-0136
[12] Luo M, Liang L, Lang L, et al. Molecular dynamics simulations of the characteristics of MoTi interfaces[J]. Computational Materials Science, 2018, 141: 293-301. DOI: 10.1016/j.commatsci.2017.09.039
[13] Bhasker-Ranganath S, Wick C D, Ramachandran B R. Computational insights into the molecular mechanisms for chromium passivation of stainless-steel surfaces[J]. Materials today chemistry, 2020, 17: 100298. DOI: 10.1016/j.mtchem.2020.100298
[14] Kumagai T, Takahashi A, Takahashi K, et al. Velocity of mixed dislocations in body centered cubic iron studied by classical molecular dynamics calculations[J]. Computational Materials Science, 2020, 180: 109721. DOI: 10.1016/j.commatsci.2020.109721
[15] Wang H, Han E. Ab initio molecular dynamics simulation on interfacial reaction behavior of Fe-Cr-Ni stainless steel in high temperature water[J]. Computational Materials Science, 2018, 149: 143-152. DOI: 10.1016/j.commatsci.2018.03.025
[16] 吕昭平, 雷智锋, 黄海龙, 等. 高熵合金的变形行为及强韧化[J]. 金属学报, 2018, 54(11): 1553-1566. DOI: 10.11900/0412.1961.2018.00372 LV Zhaoping, LEI Zhifeng, HUANG Hailong, et al. Deformation behavior and strengthening and toughening of high entropy alloys[J]. Acta Metallica Sinica, 2018, 54(11): 1553-1566(in Chinese). DOI: 10.11900/0412.1961.2018.00372
[17] Wu B, Dong H, Li P, et al. Vacuum diffusion bonding of TC4 titanium alloy and T2 copper by a slow cooling heat treatment[J]. Journal of Materials Processing Technology, 2022, 305: 117595. DOI: 10.1016/j.jmatprotec.2022.117595
[18] 佘武昌. CoSb_3/Ti界面原子扩散的分子动力学模拟[D]. 武汉理工大学, 2012. SHE Wuchang. Molecular Dynamics simulation of atomic diffusion at CoSb_3/Ti interface [D]. Wuhan University of Technology, 2012. (in Chinese)
[19] 张清东, 李硕, 张勃洋, 等. 金属轧制复合过程微观变形行为的分子动力学建模及研究[J]. 金属学报, 2019, 55(7): 919-927. DOI: 10.11900/0412.1961.2018.00524 ZHANG Qingdong, LI Shuo, ZHANG Boyang, et al. Molecular dynamics Modeling and study of microscopic deformation behavior in Metal rolling Composite Process[J]. Acta Metallica Sinica, 2019, 55(7): 919-927(in Chinese). DOI: 10.11900/0412.1961.2018.00524
[20] 罗龙, 王宝峰, 李丽荣. 铜/铝热轧扩散复合界面扩散的分子动力学模拟[J]. 热处理技术与装备, 2011, 32(2): 55-60. DOI: 10.3969/j.issn.1673-4971.2011.02.016 LUO Long, WANG Baofeng, LI Lirong. Molecular Dynamics Simulation of interfacial diffusion of Copper/aluminum hot rolling diffusion composite[J]. Heat Treatment Technology and Equipment, 2011, 32(2): 55-60(in Chinese). DOI: 10.3969/j.issn.1673-4971.2011.02.016
[21] Asche T S, Behrens P, Schneider A M. Validation of the COMPASS force field for complex inorganic–organic hybrid polymers[J]. Journal of Sol-Gel Science and Technology, 2017, 81: 195-204. DOI: 10.1007/s10971-016-4185-y
[22] 殷开梁, 邹定辉, 杨波, 等. Materials Studio 软件涉及力场中氢键的研究[J]. 计算机与应用化学, 2006, 23(12): 1335-1340. DOI: 10.3969/j.issn.1001-4160.2006.12.037 YIN Kailiang, ZOU Dinghui, YANG Bo, et al. Materials Studio software involved in hydrogen bond research in force field[J]. Computer and Applied Chemistry, 2006, 23(12): 1335-1340(in Chinese). DOI: 10.3969/j.issn.1001-4160.2006.12.037
[23] Li Y, Wang S, He E, et al. The effect of sliding velocity on the tribological properties of polymer/carbon nanotube composites[J]. Carbon, 2016, 106: 106-109. DOI: 10.1016/j.carbon.2016.04.077
[24] 肖继军, 谷成刚, 方国勇, 等. TATB 基 PBX 结合能和力学性能的理论研究[J]. 化学学报, 2005, 63(6): 439. DOI: 10.3321/j.issn:0567-7351.2005.06.001 XIAO Jijun, GU Chenggang, FANG Guoyong, et al. Theoretical study on binding energy and mechanical properties of TATB-based PBX[J]. Acta Chimica Sinica, 2005, 63(6): 439(in Chinese). DOI: 10.3321/j.issn:0567-7351.2005.06.001
[25] 梅平. 金属键能, 熔, 沸点之间的相互关系[J]. 江汉大学学报, 1991, (3): 5-9. MEI Ping. The Relationship between metal bond energy, melting point and boiling Point[J]. Journal of Jianghan University, 1991, (3): 5-9(in Chinese).
[26] Ma Y, Zhang S, Wang T, et al. Atomic diffusion behavior near the bond interface during the explosive welding process based on molecular dynamics simulations[J]. Materials Today Communications, 2022, 31: 103552. DOI: 10.1016/j.mtcomm.2022.103552
[27] 李硕. 不锈钢/碳钢层合板轧制复合机理与规律研究[D]. 北京科技大学, 2020. LI Shuo. Research on Mechanism and Law of Stainless Steel/Carbon Steel Laminated plate rolling [D]. University of Science and Technology Beijing, 2020. (in Chinese)
[28] 范同祥, 刘悦, 杨昆明, 等. 碳/金属复合材料界面结构优化及界面作用机制的研究进展[J]. 金属学报, 2019, 55(1): 16-32. DOI: 10.11900/0412.1961.2018.00509 FAN Tongxiang, LIU Yue, YANG Kunming, et al. Research progress on optimization of interfacial structure and interaction mechanism of carbon/metal composites[J]. Acta Metalica Sinica, 2019, 55(1): 16-32(in Chinese). DOI: 10.11900/0412.1961.2018.00509
[29] Liang, L. W. , Wang, Y. J. , Chen, Y. , et al. Dislocation nucleation and evolution at the ferrite-cementite interface under cyclic loadings[J]. Acta Materialia, 2020, 186: 267-277.
[30] Zepeda-Ruiz L A, Stukowski A, Oppelstrup T, et al. Probing the limits of metal plasticity with molecular dynamics simulations[J]. Nature, 2017, 550(7677): 492-495. DOI: 10.1038/nature23472
[31] 程江伟, 张先明, 吴永全, 等. α-Fe 和 γ-Fe 长程 FS 势的分子动力学模拟[J]. 物理化学学报, 2007, 23(5): 779-785. DOI: 10.3866/PKU.WHXB20070531 CHENG Jiangwei, ZHANG Xianming, WU Yongquan, et al. Molecular Dynamics Simulation of α-Fe and γ-Fe long range FS potential[J]. Journal of Physical Chemistry, 2007, 23(5): 779-785(in Chinese). DOI: 10.3866/PKU.WHXB20070531
[32] Liu B X, An Q, Yin F X, et al. Interface formation and bonding mechanisms of hot-rolled stainless steel clad plate[J]. Journal of Materials Science, 2019, 54(17): 11357-11377. DOI: 10.1007/s10853-019-03581-x
[33] 李岩, 刘琪, 李聚才, 等. 钛/铝爆炸焊接界面扩散行为分子动力学模拟(英文)[J]. 稀有金属材料与工程, 2023, 52(6): 2017-2023. DOI: 10.12442/j.issn.1002-185X.20221003 LI Yan, LIU Qi, LI Jucai, et al. Molecular Dynamics Simulation of interfacial diffusion behavior in Titanium/Aluminum explosive welding[J]. Rare Metal Materials and Engineering, 2023, 52(6): 2017-2023(in Chinese). DOI: 10.12442/j.issn.1002-185X.20221003
[34] Zhang T T, Wang W X, Zhou J, et al. Molecular dynamics simulations and experimental investigations of atomic diffusion behavior at bonding interface in an explosively welded Al/Mg alloy composite plate[J]. Acta Metallurgica Sinica (English Letters), 2017, 30: 983-991. DOI: 10.1007/s40195-017-0628-x
[35] 刘鑫, 帅美荣, 李海斌, 等. 微张力对不锈钢/碳钢界面复合质量及原子扩散的影响[J]. 材料导报, 2022, 36(23): 127-132. LIU Xin, SHUAI Meirong, LI Haibin, et al. Effects of Micro-tension on composite mass and atomic diffusion of stainless steel/Carbon steel[J]. Materials Review, 202, 36(23): 127-132. (in Chinese)
[36] 郭星星, 帅美荣, 楚志兵, 等. 不锈钢复合钢筋近界面微观组织演变及元素扩散动力学[J/OL]. 金属学报: 1-22[2024-04-19]. GUO Xingxing, SHUAI Meirong, CHU Zhibing, et al. Near-interface microstructure evolution and element diffusion kinetics of stainless steel composite bars [J/OL]. Journal of metal: 1-22 [2024-04-19]. (in Chinese)
-
-
不锈钢/碳钢复合材料因其优异的机械性能和耐腐蚀性,被广泛应用于核能核电、石油化工、海洋工程以及装甲制造等高端国防领域。与爆炸焊、堆焊等生产方法相比,热轧复合具有结合界面完整、生产效率高、安全环保等优势,是目前层状金属复合材料的主要生产方法。
深入探究不锈钢/碳钢金属界面原子扩散行为以及相变的发生发展规律,对于提升金属间冶金结合质量、实现产品性能调控具有重要意义。本文利用分子动力学模拟软件Materials Studio,建立了COMPASS力场下的不锈钢FCC-Fe和碳钢BCC-Fe晶胞模型;在热压缩高温保温和连续压缩两个阶段分别采用NVT和NPT系综,保温温度1423K,压应力分别为2GPa和4GPa;通过研究界面微观结构、均方位移分布、径向分布函数和界面元素分布模拟非共格金属界面结构演变行为。结果表明,在保温阶段,受高温影响,碳钢侧晶体发生BCC-Fe→FCC-Fe转变,空间群由P1向FM-3M的转变过程为无序长程扩散;然而不锈钢侧FCC-Fe晶体受到Cr、Ni等合金元素的固溶强化作用,晶体结构没有发生明显变形,此时界面之间存在明显的晶界。在加载阶段,随着BCC-Fe晶体相变产生的内能增加,不锈钢侧面心立方固溶体组成的超级胞滑移系开始启动,不锈钢参与到变形之中,两侧原子相互嵌入,形成统一的面心立方晶体;且随着压力增加,界面结构以最密排的(111)晶面为单位产生大量的滑移和错排,两组元原子能够发生有效的扩散迁移,界面扩散层的厚度显著增加。最后在热力模拟机上开展高温压缩试验,对不同工况下材料的宏观变形与微观元素扩散机理开展研究,高温压缩实验结果与分子动力学微观结果演变规律完全一致,从而也证明了本文中所采用的Materials Studio中的COMPASS力场和NVT、NPT系综是完全适用于模拟非共格异质金属界面微观结构变化和元素扩散迁移行为等的变化规律,以此能够揭示金属界面微观复合机理,对于此类金属复合轧制工艺优化和性能调控具有重要意义。
2GPa压应力下的界面结构模型

4GPa压应力下的界面结构模型



计量
- 文章访问数: 187
- HTML全文浏览量: 105
- PDF下载量: 7
