

Interfacial and hygrothermal properties of high modulus carbon fiber composites modified with graphene nanoplates
-
摘要:
高模量碳纤维(HMCF)复合材料的界面性能一直是影响其应用的关键所在,对HMCF进行表面改性则是实现界面性能提高的重要途径之一。采用电泳沉积法将石墨烯纳米片(GNP)引入到HMCF表面,通过SEM、XPS对改性前后的HMCF表面形貌、表面化学状态进行了表征,测试了改性前后HMCF增强环氧树脂复合材料的层间剪切强度(ILSS)和吸湿率。结果表明,HMCF表面电泳沉积GNP,不仅可以改善其增强环氧树脂复合材料的界面性能,同时也能降低复合材料的吸湿率。在GNP浓度为0.5 mg/mL,电泳沉积电压为10 V时,复合材料层间剪切性能提高了8.8%,达到75.6 MPa;90℃、80%相对湿度(RH)环境下存放30天后,与未改性样品相比,其吸湿率降低了9.5%,存放60天后层间剪切性能仍保持在67.8 MPa。改性高模量碳纤维复合材料界面性能与耐湿热性能均得以提高。
Abstract:The interfacial properties of high-modulus carbon fiber (HMCF) composites have always been the key to their applications, and surface modification of HMCF is one of the important ways to improve the interfacial properties. Graphene nanosheets (GNP) were introduced into the surface of HMCF by electrophoretic deposition, and the surface morphology and surface chemical state of HMCF before and after modification were characterized by SEM and XPS, and the interlaminar shear strength (ILSS) and moisture absorption rate of HMCF reinforced epoxy resin composites before and after modification were tested. The results show that the surface electrophoretic deposition of GNP by HMCF can not only improve the interfacial properties of the reinforced epoxy resin composites, but also reduce the moisture absorption rate of the composites. When the GNP concentration was 0.5 mg/mL and the electrophoretic deposition voltage was 10 V, the interlaminar shear performance of the composites increased by 8.8% to 75.6 MPa, and the moisture absorption rate was reduced by 9.5% compared with the unmodified sample after 30 days of storage at 90℃ and 80% relative humidity (RH), and the interlayer shear performance remained at 67.8 MPa after 60 days of storage. Both interfacial and hygrothermal properties of the modified HMCF comosites have been improved.
-
锂离子电池 (LIB) 因其比容量高、适宜的工作电压、循环寿命长和体积小等特点得到了广泛关注[1]。负极作为LIB的重要组成部分之一,其性能对电池整体的各项指标有着重要的影响[2]。当前占据市场上最大份额的石墨负极材料比容量已接近上限,其层状结构限制了快速充电应用,且较低的对锂电位易导致析锂,存在安全隐患等问题[3]。与此相比,SiOx(x≈1)因其高锂扩散系数、高比容量(
2100 mA·h·g−1)、出色的快速充放电性能和适当的工作电位等特性备受青睐,被认为是最具潜力的下一代负极材料。然而,SiOx中的SiO2在首次嵌锂过程中会与活性锂离子生成不可逆的锂硅酸盐和Li2O等物质,这些不可逆相消耗了部分活性锂离子,导致了首次库仑效率(ICE)的降低[4-6]。负极材料的ICE直接影响正极材料的容量以及电池的整体设计[7];目前,产业化生产中主要通过预锂化[8-9]和预镁化[10-12]等方法来解决SiOx低ICE的问题。预镁化SiOx的ICE与电化学性能相比于预锂化SiOx较差,但由于镁源的价格更低,可降低生产成本。但近年来,随着锂源价格的下跌,汽车企业对于可商用的预锂化SiOx需求更为迫切。预锂化技术主要分为电化学预锂化、化学预锂化、物理预锂化三类[13];化学预锂化可进一步细分为液相化学预锂化和固相预锂化。电化学预锂化主要通过改变电流以及电压来控制预锂化进程,例如:外电路控制法[14],然而预锂化后的负极具有较高的化学反应活性,无法在空气中稳定保存,限制了其实际应用。液相化学预锂化是指利用还原强度较强的含锂试剂,通过氧化还原反应将活性锂离子转移到SiOx上。例如:浸泡法,通过将极片浸泡在含锂的有机溶液(锂-3,3',4,4'-四甲基联苯、Li-4,4'-二甲基联苯/四氢呋喃(THF)等)中便可得到较高的ICE[15-16]。然而,预锂化过程中会产生包含锂盐和其他化学物质的废液,易造成环境污染,预锂化过程需要在特制的反应容器中进行,对设备要求较高,上述问题造成了该项技术较难实现工业化应用。稳定锂金属粉末(SLMP)预锂化[17-18]为物理预锂化中的一种常用方法,目前主要由熔融分散和液滴乳化技术制备,首先将SLMP分散于负极上,后续再进行加压活化。SLMP预锂化的工艺简单,无死锂的残留,但该方法在制作过程中要求较高的环境管控,也存在一定的安全风险。上述问题使得电化学预锂化、物理预锂化以及液相化学预锂化后的SiOx较难规模化应用。
固相预锂化主要通过将锂源(LiOH、Li2CO3、LiH等)与SiOx进行简单的干法混合热处理,便可以得到预锂化效果优异的硅氧材料,如:较高的ICE、良好的循环稳定性等。且工艺方法简单,对环境友好,安全风险相对较低。此外,预锂化后的SiOx在空气中的稳定性较高,在湿度较低的环境下可以稳定保存。由于上述优势,固相预锂化技术已成为SiOx产品ICE提升主要的产业化技术路线。
固相预锂化是近年来硅氧负极材料研究的热点,但总结该技术成果的论文较少,没有对固相预锂化的原理、反应锂源、改进方法、热处理参数及产业化问题等角度进行全方位的总结,而本文主要介绍了固相预锂化SiOx的原理,探讨了不同锂源在固相预锂化SiOx过程中的效果差异,以及归纳了固相预锂化效果优化的方法。同时,总结了当前SiOx固相预锂化存在的问题,并介绍了产业界采取的解决方案。最后,展望了固相预锂化SiOx在锂离子电池中的应用趋势,使读者能够快速了解该领域的发展现状。(由于硅氧化物尚未完全统一称谓,本文选择以 SiOx为硅氧材料的表述)
1. 固相预锂化概述
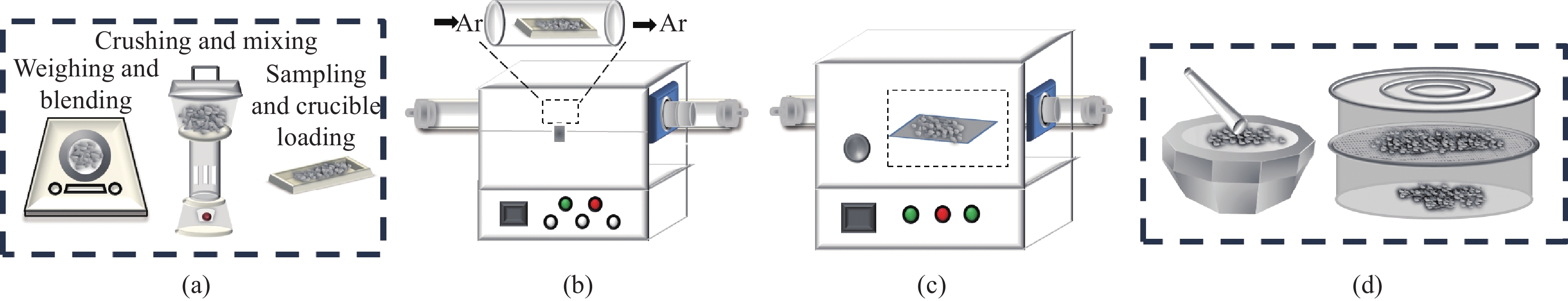
目前,固相预锂化SiOx的主要工艺步骤如图1所示,主要分为称重混料、热处理、包覆处理、研磨过筛4个步骤。热处理过程是固相预锂化中最为关键的步骤之一,不同的热处理温度以及升温速率会影响硅晶粒的大小、预锂化的完全性等,从而影响电池的ICE、循环稳定性等。因此,合理的热处理参数设置对SiOx的预锂化效果非常重要。包覆处理属于预锂化SiOx的后处理工艺,由于固相预锂化后的SiOx表面的残锂与裸露的Li2SiO3、Li4SiO4、Li2Si2O5等,在制浆工艺中会与水接触使得浆料pH值上升,会导致产气问题以及与粘结剂反应等问题,从而影响电池的性能。对于固相预锂化后的SiOx高碱性带来的一系列问题,当前产业界主要通过包覆处理的方法解决,如:碳包覆、锂硅酸盐包覆、聚合物包覆等,可以较好地解决浆料高碱性的问题。但包覆处理工艺方法增加了工艺流程的复杂度,提高了生产成本。当前产业界对于固相预锂化后的SiOx高碱性问题的解决方法仍然在探索改进中。
![]() 图 1 SiOx固相预锂化生产的主要工艺步骤:(a)称重混料;(b)热处理;(c)包覆处理;(d)研磨过筛Figure 1. Main process steps of SiOx solid phase prelithiation production: (a) Weighing and mixing; (b) Heat treatment; (c) Coating treatment; (d) Grinding and sieving
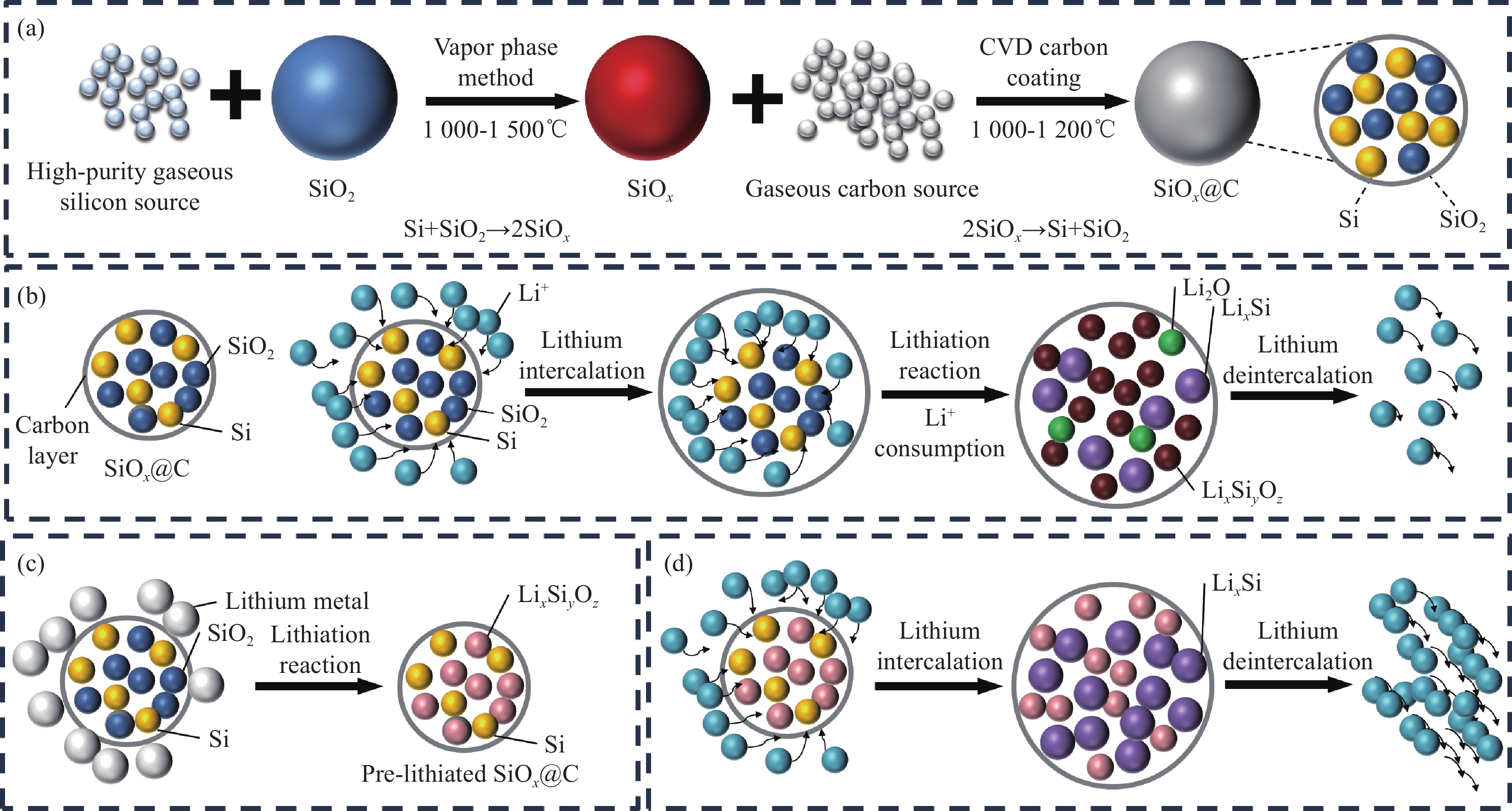
图 1 SiOx固相预锂化生产的主要工艺步骤:(a)称重混料;(b)热处理;(c)包覆处理;(d)研磨过筛Figure 1. Main process steps of SiOx solid phase prelithiation production: (a) Weighing and mixing; (b) Heat treatment; (c) Coating treatment; (d) Grinding and sieving此外,固相预锂化ICE提升的原理与Jang等[15]提出的通过补充额外的锂元素以降低SiOx在首次嵌锂中活性锂离子消耗的概念不同。由于商业化的SiOx主要与碳复合形成核壳结构的碳包覆微米级材料,与石墨按照一定比例的混合作为负极材料使用[19-20]。因此在碳包覆的工艺流程中,SiOx在高温(>
1000 ℃)下会歧化成由Si和SiO2纳米颗粒组成的复合材料[21-24](图2(a)),而SiO2会在首次嵌锂过程中消耗活性锂离子(图2(b)),从而降低ICE。固相预锂化主要是指在较高温度条件下,使锂源(LiH、LiOH、Li2CO3等)与SiOx中因高温歧化产生的SiO2提前发生锂化反应,生成一系列稳定性较好的锂硅酸盐,最大程度上降低SiOx首次嵌锂过程中活性锂离子的消耗,从而提升ICE。图2(c)为锂金属固相预锂化SiOx的示意图,通过锂金属与SiO2发生的反应,提前消耗SiOx中的SiO2,生成了一系列稳定的锂硅酸盐(Li2SiO3、Li4SiO4)。避免了首次嵌锂过程中SiO2与活性锂离子反应,生成不可逆的锂硅酸盐以及氧化锂,造成活性锂离子的损失(图2(d))。![]() 图 2 (a)化学气相沉积(CVD)碳包覆SiOx示意图;(b) SiOx@C首次脱嵌锂过程示意图;(c)锂金属固相预锂化SiOx@C示意图 ;(d)预锂化SiOx@C的脱嵌锂过程示意图Figure 2. (a) Schematic diagram of chemical vapor deposition (CVD) carbon-coated SiOx; (b) First lithium intercalation and deintercalation process of SiOx@C; (c) Schematic diagram of lithium metal solid-phase prelithiation of SiOx@C; (d) Lithium intercalation and deintercalation process of prelithiated SiOx@C
图 2 (a)化学气相沉积(CVD)碳包覆SiOx示意图;(b) SiOx@C首次脱嵌锂过程示意图;(c)锂金属固相预锂化SiOx@C示意图 ;(d)预锂化SiOx@C的脱嵌锂过程示意图Figure 2. (a) Schematic diagram of chemical vapor deposition (CVD) carbon-coated SiOx; (b) First lithium intercalation and deintercalation process of SiOx@C; (c) Schematic diagram of lithium metal solid-phase prelithiation of SiOx@C; (d) Lithium intercalation and deintercalation process of prelithiated SiOx@C2. 不同锂源固相预锂化SiOx研究
2.1 锂金属固相预锂化
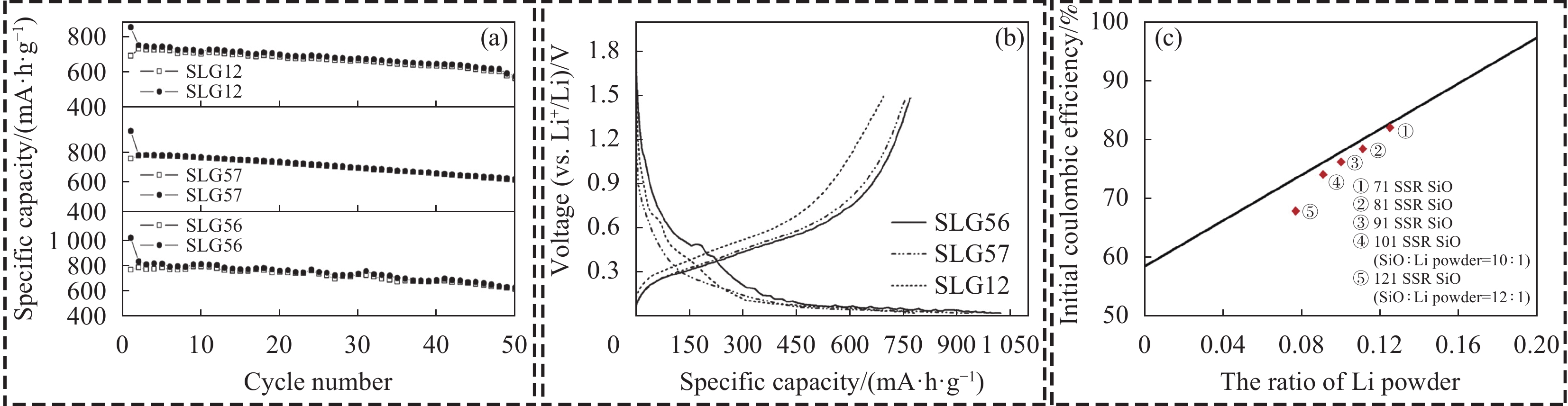
最早的固相预锂化方法以锂金属作为锂源。Yang等[25]通过高能机械铣削(HEMM)工艺混合SiOx与锂粉,添加10wt%的石墨来抑制因锂金属熔化而产生的结块,最后在真空下进行热处理,得到Si@Li4SiO4结构的复合材料。半电池ICE可达81%,比容量为770.4 mA·h·g−1,ICE远高于裸SiOx (58.52%)。正硅酸锂(Li4SiO4)的生成可有效的吸收硅颗粒的体积变化,为Li+嵌入/脱出硅颗粒提供有利的环境[26];在此基础之上,Yang等[27]探究了SiOx/Li摩尔比(SLG表示)为5/6、5/7和1/2对SiOx固相预锂化的影响。随着锂粉量的增加,比容量从
1027 mA·h·g−1 (SLG56)下降到853 mA·h·g−1 (SLG12),ICE从74.9%增加到81.3%;且在第50次循环时的可逆容量分别为622.4、614.2和570.1 mA·h·g−1,容量保持率分别为80.9%、81.2%和82.1% (图3(a))。循环稳定性的提升主要由于锂粉量增加,生成了更多的Li4SiO4相。此外,SLG56在0.6 V以上的不可逆容量损失大于SLG12和SLG57 (图3(b)),过量的锂粉可以补偿固态电解质界面(SEI)形成引起的活性锂离子的消耗。![]() Figure 3. Cycling performance (a) and initial charge-discharge curves (b) of SiOx with different SiOx/Li molar ratios (SLG)[27]; (c) Function relationship of the proportion of initial coulombic efficiency (ICE) and the ratio of lithium powder[8]SLG12, SLG57, SLG56—SiOx/Li molar ratios 1/2, 5/7, 5/6
Figure 3. Cycling performance (a) and initial charge-discharge curves (b) of SiOx with different SiOx/Li molar ratios (SLG)[27]; (c) Function relationship of the proportion of initial coulombic efficiency (ICE) and the ratio of lithium powder[8]SLG12, SLG57, SLG56—SiOx/Li molar ratios 1/2, 5/7, 5/6Yom等[8]同样探究了SiOx/Li质量比为7∶1、8∶1、9∶1 (统称71SSR、81SSR、91SSR)对锂金属预锂化SiOx的影响,解释了锂粉量的增加导致电池比容量降低、ICE提升的原因。随着锂粉的比重减少,ICE逐渐下降(图3(c))。造成该现象的原因可能与锂化反应有关,在81SSR SiOx和91SSR SiOx电极的情况下,锂粉的量不足以完全消耗SiO2形成更多的不可逆相。因此,在首次嵌锂过程中,剩余的SiO2和锂离子之间的电化学反应产生了额外的不可逆相,消耗了更多的活性锂离子,降低了ICE。而71SSR SiOx的锂含量较高,消耗了更多的SiO2,减少了活性锂离子的损失,提高了ICE。且残锂的存留会使得硅含量降低,导致可逆容量的损失。由于锂金属没有容量,当与活性物质混在一起时,会导致活性物质质量的占比量偏小。比容量的公式为容量(mA·h)/质量(g),而预锂化后比容量的公式变为容量(mA·h)/[活性物质质量(g)+残锂质量(g)],由于活性物质的比例不变,因此预锂化后材料的实际比容量偏低。
锂金属固相预锂化SiOx能够带来明显的ICE提升效果,但其锂含量高,预锂化后较多的残锂存留量使得电池的比容量损失较大。除此之外,锂金属在空气中的高反应活性限制了其实际应用,存在较大的安全隐患,难以大规模化生产。
2.2 锂化合物固相预锂化
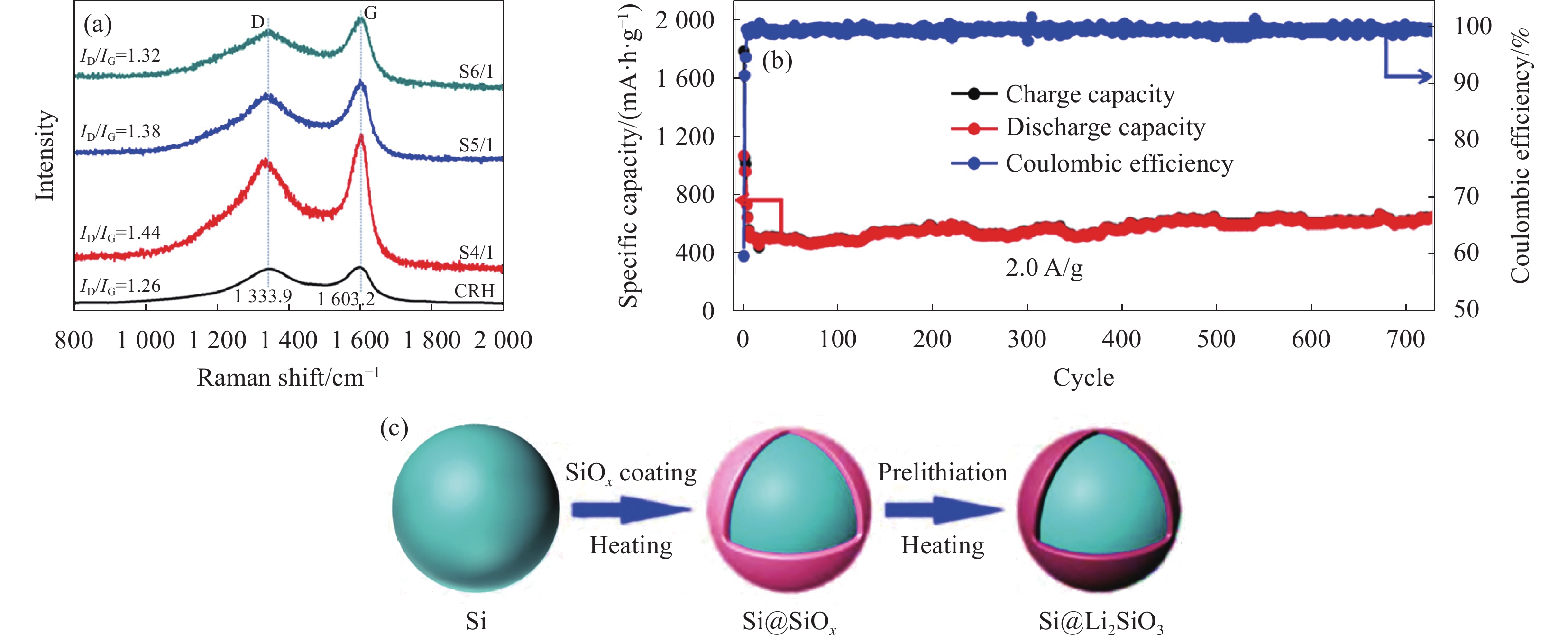
在此情况下,LiOH·H2O、Li2CO3、LiBH4、LiH等具备还原性的锂源成为了固相预锂化的重要研究对象。Veluchamy等[28]以LiOH·H2O作为锂源,在550℃下对裸SiOx进行固相预锂化。但预锂化后的ICE相对于裸SiOx无明显提升,可逆容量仅有387 mA·h·g−1,但循环稳定性提升明显。较好的循环稳定性主要是由于预锂化后Li4SiO4的生成。Yi等[29]使用Li2CO3作为锂源,与多孔碳化稻壳的SiOx颗粒按照4∶1、5∶1和6∶1质量比进行混合热处理(分别命名为S4/1、S5/1和S6/1),获得了多孔SiOx@Li2SiO3/C复合结构材料。随着Li2CO3掺杂量的提高,ID/IG比值(拉曼光谱中D峰(C原子晶格的缺陷)与G峰(C原子sp2杂化的面内伸缩振动)的比值)逐渐增大,表明掺杂Li2CO3会降低材料的石墨化程度,增加缺陷程度,可为材料提供更多的Li+储存位点(图4(a))。在后续的半电池性能测试中,所获得的SiOx@Li2SiO3/C复合材料(S5/1)在0.1 A·g−1下具有825 mA·h·g−1的可逆容量,ICE提升到约80%;在2 A·g−1的电流密度下,其比容量在720次循环后可保持在约620 mA·h·g−1 (图4(b))。为了进一步提高预锂化后的ICE,Zhu等[30]用熔点(268℃)较低的硼氢化锂(LiBH4)作为锂源,图4(c)为Si@Li2SiO3制备路线示意图。半电池表现出高达89.1%的ICE,在3 A·g−1的电流密度下,循环
1000 圈后仍有959 mA·h·g−1。但其预锂化工艺复杂度较高,且LiBH4化学性质较为活泼,遇潮湿空气和水易发生反应放出易燃的氢气,存在较大的安全隐患,限制了其实际应用。![]() S6/1, S5/1, S4/1—Mass ratio of porous carbonized rice husk to SiOx (6:1, 5:1, 4:1); CRH—Rice husk after direct carbonization; ID/IG—Ratio of the D peak (defects in the carbon atom lattice) to the G peak (in-plane stretching vibration of sp² hybridized carbon atoms) in Raman spectroscopy
S6/1, S5/1, S4/1—Mass ratio of porous carbonized rice husk to SiOx (6:1, 5:1, 4:1); CRH—Rice husk after direct carbonization; ID/IG—Ratio of the D peak (defects in the carbon atom lattice) to the G peak (in-plane stretching vibration of sp² hybridized carbon atoms) in Raman spectroscopy为了简化SiOx固相预锂化的工艺,提高ICE。Chung等[9]选用热稳定性较高的LiH作为锂源,通过最简单的混合热处理的固相预锂化方法得到了较高ICE的SiOx,工艺步骤如图5(a)所示。Li/Si=0.67 (摩尔比)预锂化后的SiOx电性能最为优异,ICE从59.3%提升至90.5%,可逆容量为
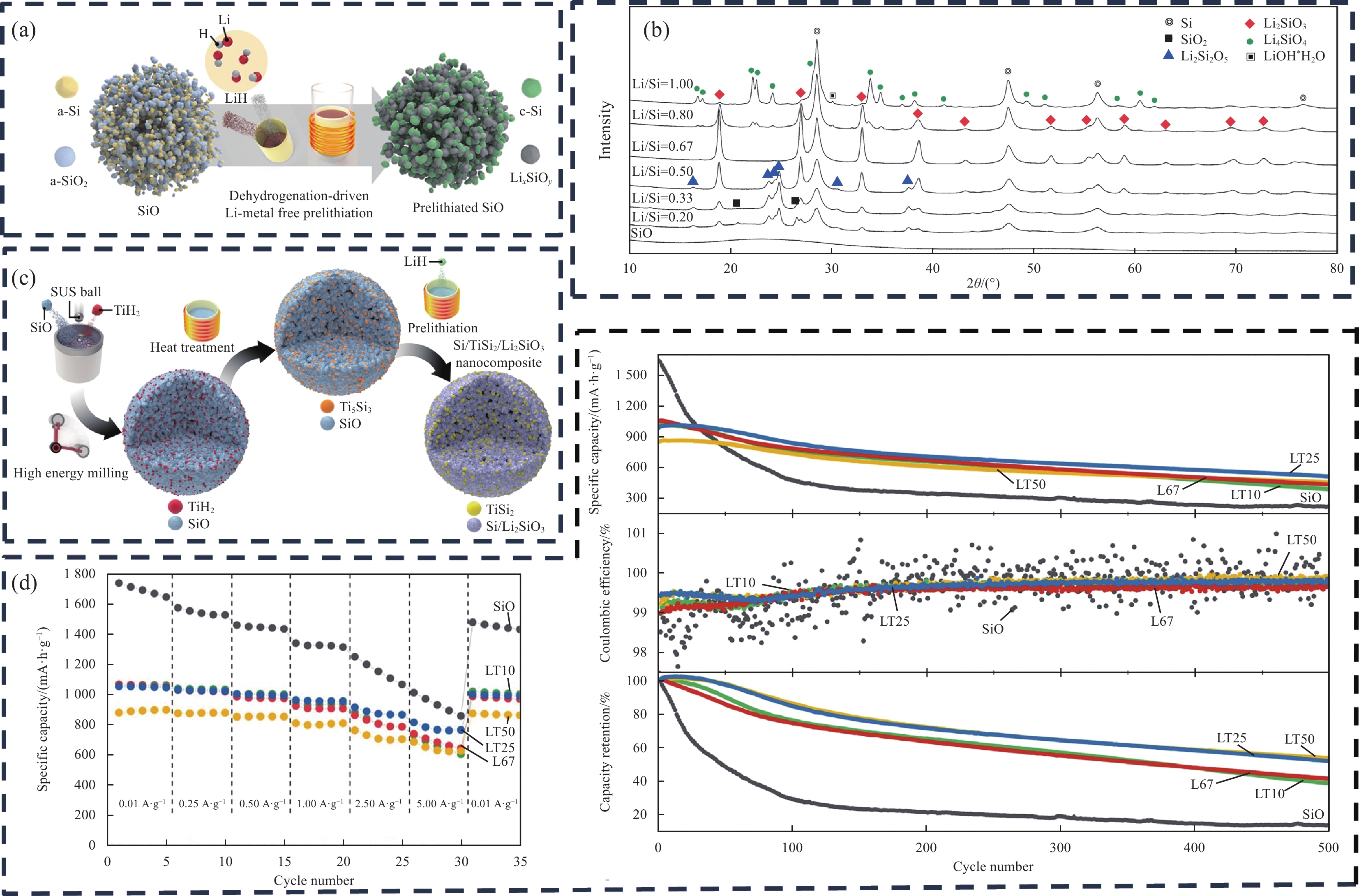
1203 mA·h·g−1,其预锂化提升效果比上述预锂化方法更为明显。全电池(37 mA·h)的能量密度提高了50%,并具有超过800圈的优异循环性能。目前为止,通过LiH固相预锂化SiOx,其ICE提升效果是最为显著的。Li/Si=0.67时,Li2SiO3相生成的衍射峰是最为明显的(图5(b)),且Si活性相和Li2SiO3非活性缓冲相的拓扑排列是增强预锂化后的SiOx长期循环稳定性和体积变化的关键[31],这也是Li/Si=0.67预锂化SiOx性能优异的原因。![]() Figure 5. Schematic diagram of LiH prelithiated SiOx (a) and XRD patterns after prelithiation (b)[9]; Schematic diagram of Si/TiSi2/Li2SiO3 preparation (c) and rate performance, cycling capacity, efficiency curves (d)[32]SUS—Stainless steel ball; L67—Molar ratio of Li to Si is 0.67; LT(10/25/50): Molar ratios of Ti to Li are 0.01, 0.025, 0.050
Figure 5. Schematic diagram of LiH prelithiated SiOx (a) and XRD patterns after prelithiation (b)[9]; Schematic diagram of Si/TiSi2/Li2SiO3 preparation (c) and rate performance, cycling capacity, efficiency curves (d)[32]SUS—Stainless steel ball; L67—Molar ratio of Li to Si is 0.67; LT(10/25/50): Molar ratios of Ti to Li are 0.01, 0.025, 0.050为了进一步提升LiH预锂化SiOx负极材料的电化学性能;Jeong等[32]提出了一种通过SiOx与金属氢化物相反应制备双缓冲相嵌入Si@TiSi2@Li2SiO3@C纳米复合材料结构的方法。TiSi2对Si颗粒的体积膨胀具有较好的缓冲性能,可以增强硅基负极材料的循环稳定性,提升材料的倍率性能[33-37]。Si@TiSi2@Li2SiO3纳米复合材料及其制备示意图如图5(c)所示。相对于Si@Li2SiO3@C,Si@TiSi2@Li2SiO3@C复合材料循环性能、倍率性能有明显提升(图5(d)),ICE无明显降低,但比容量下降了近100~200 mA·h·g−1,主要是由于TiH2的加入导致活性物质的比例降低,使得比容量下降。虽然新引入的TiSi2缓冲相可明显提升LiH预锂化SiOx的循环、倍率等各方面性能,但也增加了工艺的流程复杂度。
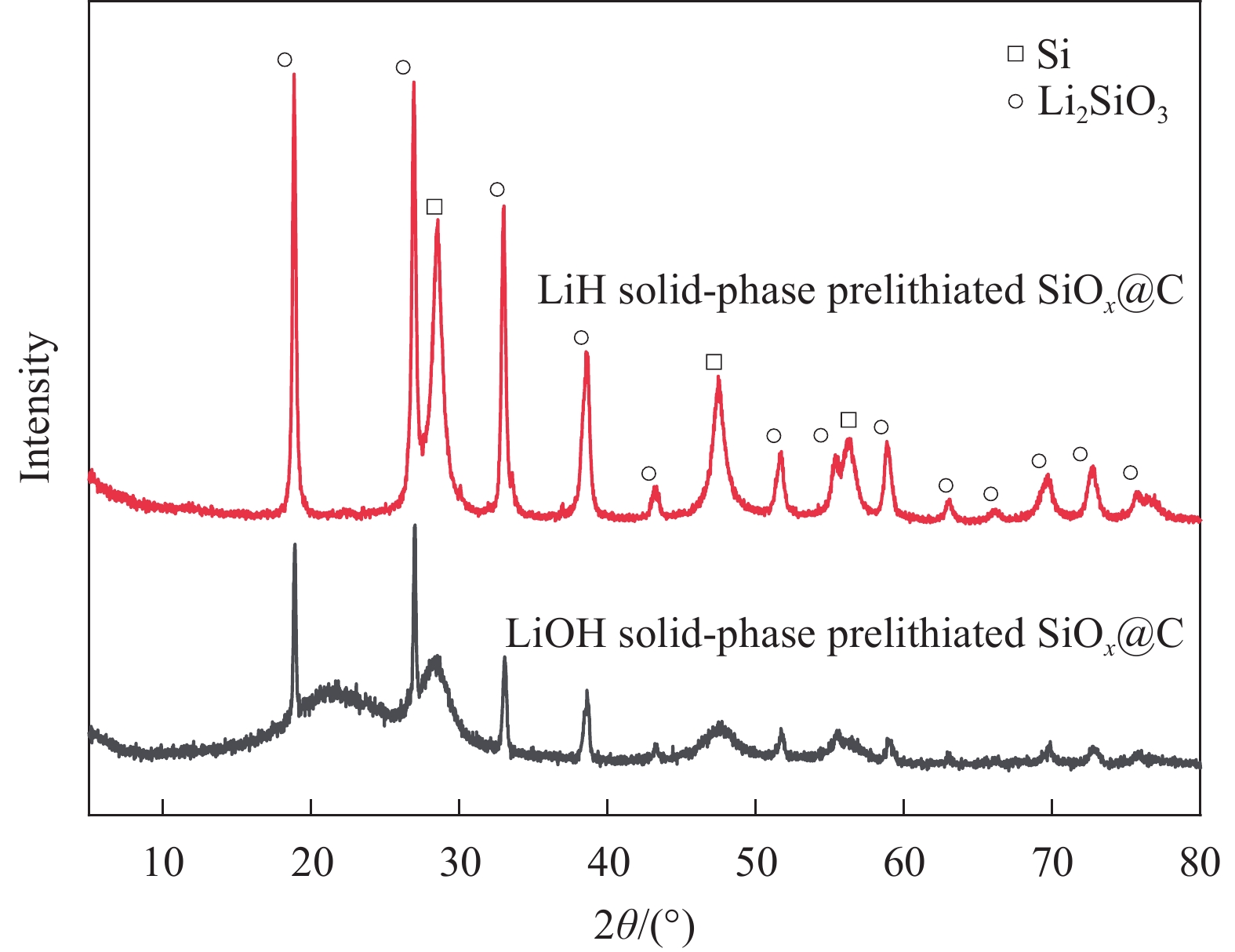
LiOH·H2O、Li2CO3、LiBH4、LiH固相预锂化效果差异分析:(1) LiOH·H2O对 SiOx的固相预锂化效果无明显提升。主要归咎于其低反应活性和高分解温度,导致与 SiOx 反应速率缓慢。且其锂含量较低,难以在预锂化过程中产生显著效果。此外,其生成水的副反应也会影响预锂化的反应进程;(2)使用Li2CO3作为锂源,ICE可提升至80%左右。相比其他锂源,Li2CO3的制备成本相对较低,在实际的中已得到广泛的应用。但与SiOx之间的反应速率适中,且固相颗粒间本身混合的均匀性差,难以使其预锂化的效果有进一步的提升;(3) LiBH4预锂化后的ICE优异,主要归咎于其较高的反应活性[38-39],在较低的温度下便能与SiOx发生反应。且锂含量高[40],能够为SiO2的消耗,提供更多的反应物来源。但反应过程中可能会生成一些副产物[41],如:乙硼烷/六氢化二硼(B2H6),且会有更多的残锂存留,需要对预锂化后的杂质进行处理,增加了工艺流程的复杂度,同样存在预锂化不均匀的问题。除此之外,LiBH4化学性质活泼,存在较大的安全隐患。尽管如此,其高 ICE 和优异的循环性能仍具一定应用价值;(4) LiH预锂化SiOx的效果最为优异,相对于LiBH4具有更高的热稳定性,在室温下能够相对稳定存在。优异的预锂化效果主要由于其可提供高纯度的锂,与SiOx之间的反应活性高,副产物相对较少,减少了对预锂化过程的影响。反应产物中的 Si 活性相和 Li2SiO3 非活性缓冲相的拓扑排列结构也增强了 SiOx 的循环稳定性。此外,LiH脱氢反应释放的氢气以及产生的能量有利于驱动相关的化学反应[42-44],提升了预锂化的效率,这也是LiH固相预锂化不同于其他锂化合物的地方。但LiH具有较高的键能,放出的热量更多,相对于其他锂源对硅晶粒尺寸的影响也更大(图6、表1 ),同样的热处理参数条件下,LiOH预锂化SiOx@C后的硅晶粒尺寸为5.4 nm,LiH预锂化后的硅晶粒尺寸为7.35 nm。且LiH的含量纯度高,预锂化后残锂更多,碱性更高,产气现象更严重,对电池工艺生产造成一定阻碍。虽然LiH预锂化SiOx存在上述问题,但其工艺简单、对环境友好、预锂化效果优异等,更具有产业化的潜力。
![]() 图 6 LiH与LiOH固相预锂化SiOx@C后的XRD图谱Figure 6. XRD patterns after LiH and LiOH solid-phase prelithiated SiOx@C表 1 LiH与LiOH固相预锂化SiOx@C后的硅晶粒尺寸与热处理参数Table 1. Silicon grain sizes and heat treatment parameters after solid-phase prelithiation of SiOx@C with LiH and LiOH
图 6 LiH与LiOH固相预锂化SiOx@C后的XRD图谱Figure 6. XRD patterns after LiH and LiOH solid-phase prelithiated SiOx@C表 1 LiH与LiOH固相预锂化SiOx@C后的硅晶粒尺寸与热处理参数Table 1. Silicon grain sizes and heat treatment parameters after solid-phase prelithiation of SiOx@C with LiH and LiOHHeating rate/(℃·min−1) Heating temperature/℃ Heat-up time/h Silicon grain size/nm 3 750 8 LiH solid-phase
prelithiated SiOx@CLiOH solid-phase
prelithiated SiOx@C7.35 5.4 3. 固相预锂化效果优化研究
3.1 锂源湿法包覆SiOx处理
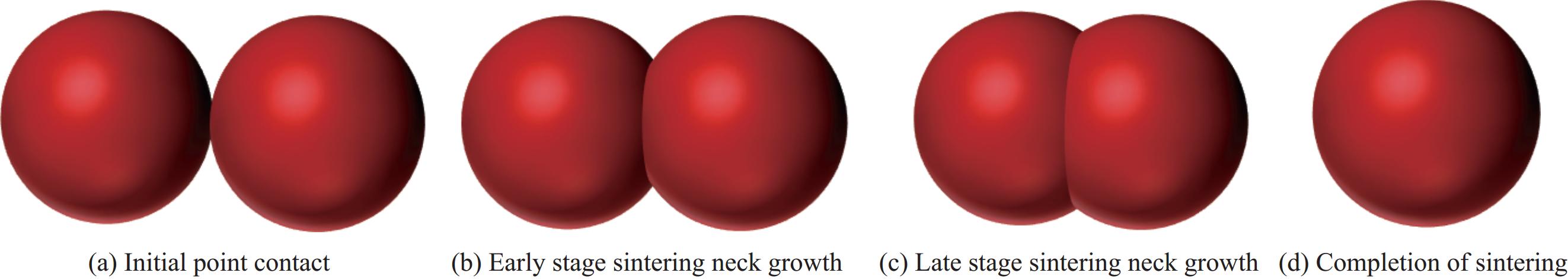
以锂化合物(Li2CO3、LiBH4、LiH)作为SiOx固相预锂化的锂源,有着较为优异的预锂化效果(ICE>80%)。但固相预锂化中颗粒间混合的均匀性差,接触位点小[45](图7),存在预锂化不均匀的问题,难以使其ICE得到进一步的提升。为解决预锂化不均匀的问题,研究者们通过将锂源湿法处理,以此实现对SiOx的均匀包覆,从而提升预锂化的均匀性。
![]() 图 7 固相颗粒热处理的4个过程Figure 7. Four processes of thermal treatment of solid-phase particles
图 7 固相颗粒热处理的4个过程Figure 7. Four processes of thermal treatment of solid-phase particlesXie等[46]将锂金属与碳酸盐溶解在聚乙烯醇(PVA)溶液中,后将SiOx与沥青粉与Li2CO3/PVA溶液混合,提高了Li2CO3对SiOx的包覆均匀性;热处理、酸洗后,制备得到了Si/SiOx/Li2SiO3@C多孔复合材料(图8(a))。半电池在0.1 A·g−1下具有
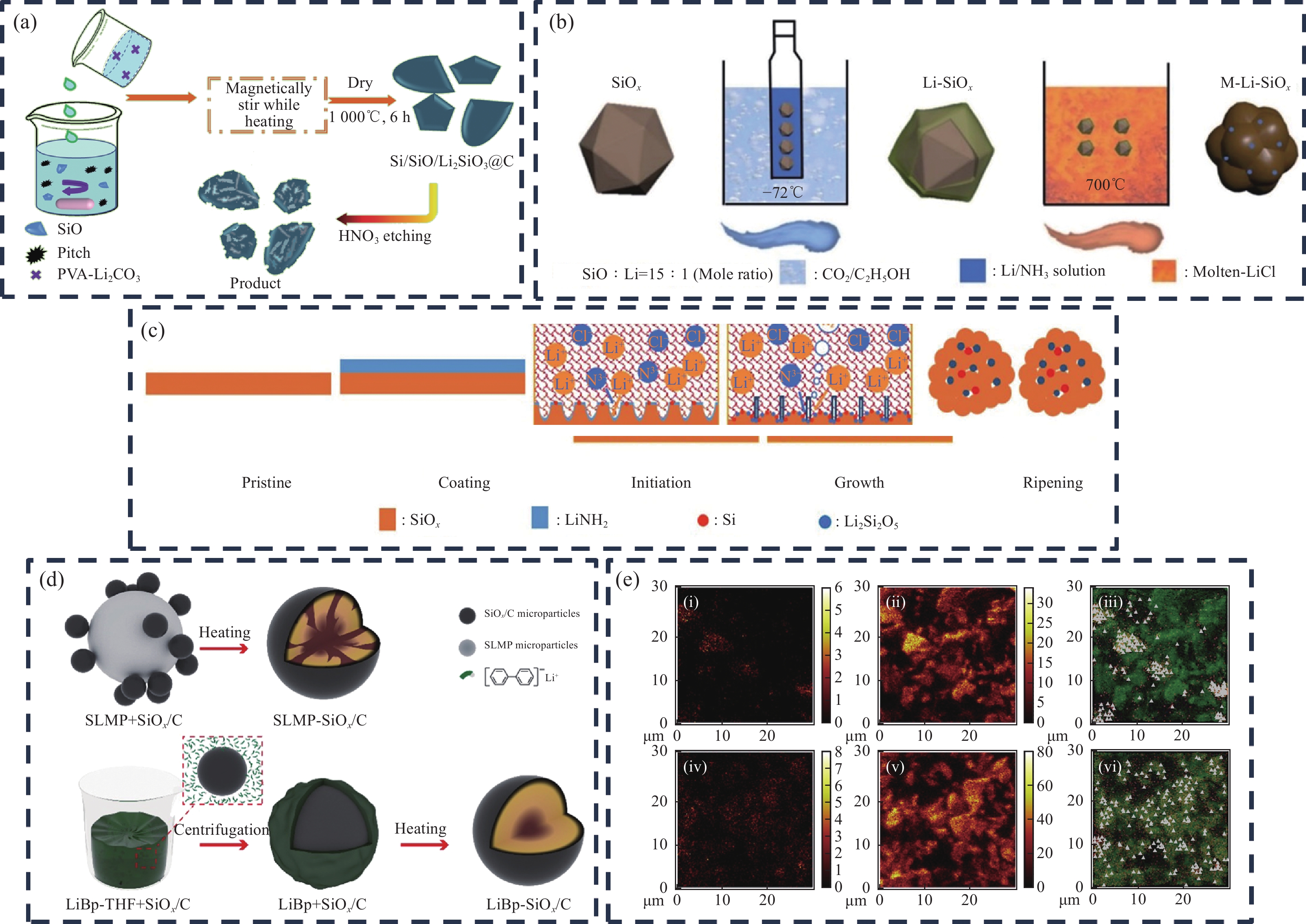
1645.47 mA·h·g−1的高比容量,但ICE仅为69.05%。为了提升ICE,Li等[47]以液态的氨基锂(LiNH2)作为锂源,用商业的SiOx、Li箔、NH3制备了Li-SiOx (SiOx@LiNH2),与无水LiCl在Ar气氛下研磨均匀并放入Al2O3舟中,最后热处理、洗涤后便得到了M-Li-SiOx粉末(图8(b))。SiOx表面涂覆的LiNH2在加热过程中会先分解产生NH3和Li3N[48-49]。而生成的Li3N可以溶解在熔融LiCl中,在界面交界处会与SiOx反应生成LixSiyOz和Si;随着反应的持续进行,最终形成其核心为SiOx,外部由Li2Si2O5、SiO2和Si组成的纳米尺寸团聚体,图8(c)为反应机制图。M-Li-SiOx半电池的ICE为88.2%,相对于裸SiOx (58.73%)提升明显,可逆容量为1013.3 mA·h·g−1。该预锂化方法相对于传统的固相预锂化其均匀性程度更高,但工艺复杂度也较高。除此之外,高温下生成Li2Si2O5时,容易发生硅的过度生长[50],使得硅晶粒增大,影响电池的长循环性能。该预锂化方法结合了低温液相涂层包覆、高温熔盐反应、固相热处理于一体的机制,可为SiOx预锂化方法的发展提供新的思路。![]() 图 8 (a) Si/SiOx/Li2SiO3@C制备示意图[46];M-Li-SiOx的制备示意图(b)以及反应过程原理图(c)[47];(d) SLMP-SiOx/C、LiBp-SiOx/C的合成过程示意图[51];(e) 飞行时间二次离子质谱(ToF-SIMS)元素分布图像(Li−((i), (iv))、Si− ((ii), (v))和Li−(红色)和Si−(绿色)物质叠加((iii), (vi));((i)~(iii)) SLMP-SiOx/C;((iv)~(vi)) LiBp-SiOx/C[51])Figure 8. (a) Schematic diagram of Si/SiOx/Li2SiO3@C preparation[46]; Preparation schematic (b) and schematic of the reaction process (c) of M-Li-SiOx[47]; (d) Schematic synthesis process of SLMP-SiOx/C, LiBp-SiOx/C[51]; (e) Time of flight secondary ion mass spectrometry (ToF-SIMS) elemental distribution images (Li− ((i), (iv)), Si− ((ii), (v)), and overlaid images of Li− (in red) and Si− (in green) ((iii), (vi)); ((i)~(iii)) SLMP-SiOx/C; ((iv)~(vi)) LiBp-SiOx/C) [51]PVA—Polyvinyl alcohol; SLMP—Stable lithium metal powder; LiBp—Lithium biphenyl; M-Li-SiOx—Multi-component prelithiated SiOx
图 8 (a) Si/SiOx/Li2SiO3@C制备示意图[46];M-Li-SiOx的制备示意图(b)以及反应过程原理图(c)[47];(d) SLMP-SiOx/C、LiBp-SiOx/C的合成过程示意图[51];(e) 飞行时间二次离子质谱(ToF-SIMS)元素分布图像(Li−((i), (iv))、Si− ((ii), (v))和Li−(红色)和Si−(绿色)物质叠加((iii), (vi));((i)~(iii)) SLMP-SiOx/C;((iv)~(vi)) LiBp-SiOx/C[51])Figure 8. (a) Schematic diagram of Si/SiOx/Li2SiO3@C preparation[46]; Preparation schematic (b) and schematic of the reaction process (c) of M-Li-SiOx[47]; (d) Schematic synthesis process of SLMP-SiOx/C, LiBp-SiOx/C[51]; (e) Time of flight secondary ion mass spectrometry (ToF-SIMS) elemental distribution images (Li− ((i), (iv)), Si− ((ii), (v)), and overlaid images of Li− (in red) and Si− (in green) ((iii), (vi)); ((i)~(iii)) SLMP-SiOx/C; ((iv)~(vi)) LiBp-SiOx/C) [51]PVA—Polyvinyl alcohol; SLMP—Stable lithium metal powder; LiBp—Lithium biphenyl; M-Li-SiOx—Multi-component prelithiated SiOxYan等[51]将锂片溶入到联苯的THF溶液中制备了锂联苯(LiBp)络合物溶液,通过加热搅拌等工艺获得了LiBp均匀吸附的SiOx/C,最后热处理得到了Si@Li2Si2O5材料。该方法也极大的提高了预锂化的均匀性(图8(d)),从飞行时间二次离子质谱(ToF-SIMS)元素分布图像亦可看出(图8(e)),Li2Si2O5呈现了均匀分布的状态。该工艺方法预锂化后的材料半电池的ICE高达90%,可逆容量为
1349 mA·h·g−1,预锂化后的SiOx/C材料效果优异;相对于上述的液态氨基锂均匀预锂化,该工艺方法简单,预锂化效果更为优异。但有机溶液的加入,增加了后处理工艺的成本。且高温下生成Li2Si2O5时,容易发生硅的过度生长的问题依旧没有解决。通过对SiOx进行湿法包覆,可在一定程度上实现对SiOx的均匀预锂化,提升预锂化效果。但其ICE的提升效果依旧难以超越LiH预锂化SiOx的干法混合热处理的方法。此外,大多数的锂化合物只能溶于有机溶液,对环境不友好,且增加了工艺复杂度,难以适应如今新能源材料行业低成本、环保的发展方向。相比之下,干法混合热处理的固相预锂化方法更具有产业化研究的价值。
3.2 偏硅酸锂相调控
Li4SiO4、Li2SiO3都是锂离子的良好导体,具有较好的缓冲效应,可抑制硅晶粒的体积膨胀,提高循环稳定性等特点,是固相预锂化中锂源(LiH、LiOH、Li2CO3等)与SiOx中SiO2的锂化反应生成的重要产物。但Li4SiO4、Li2SiO3在化学性质上以及生成所需的热处理参数条件上,均有着不同的差异,对预锂化后材料性能的提升效果也不同。
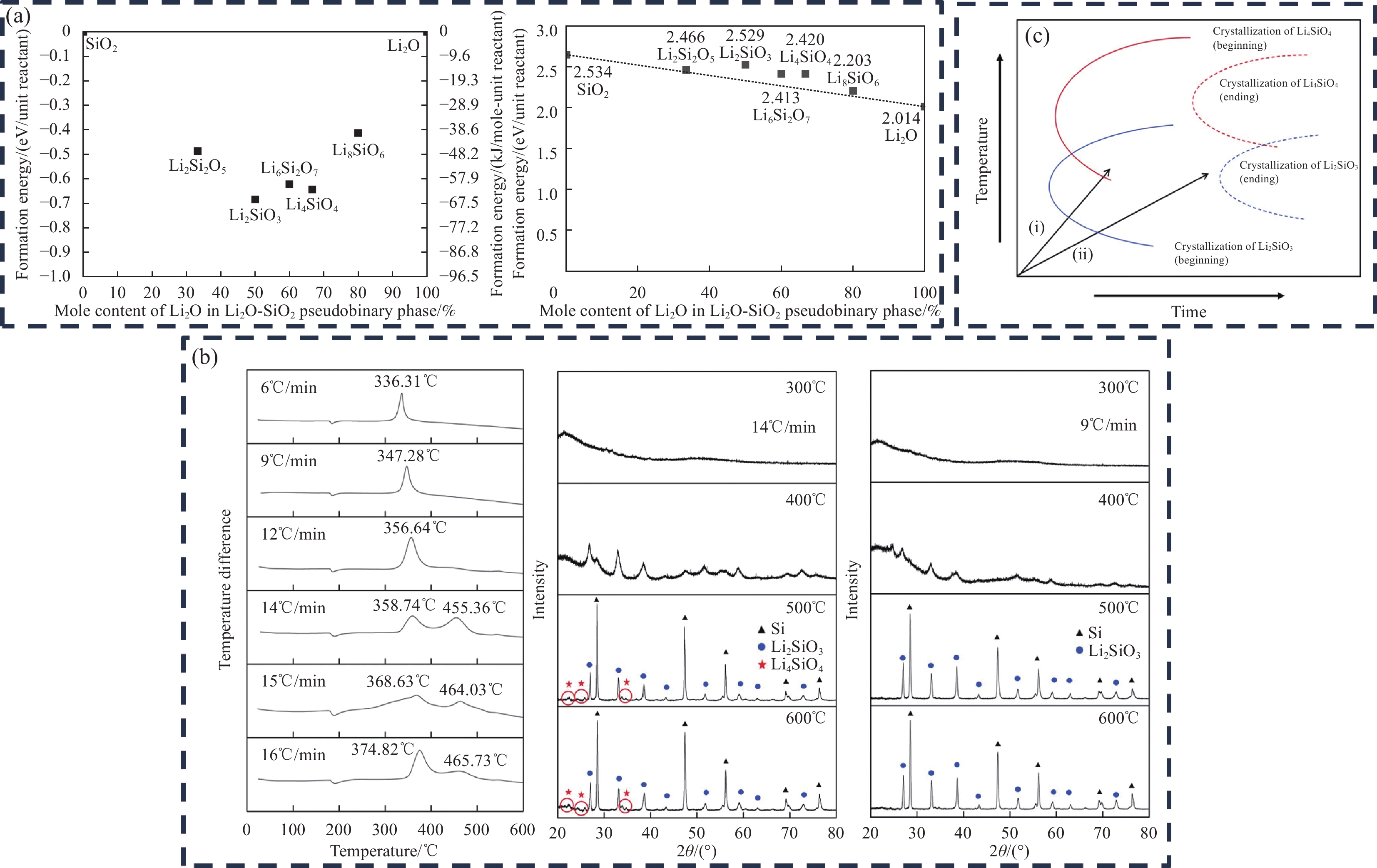
为了探究Li4SiO4与Li2SiO3的差异,Doh等[52]使用维也纳从头算模拟 (VASP)[53-54]软件包用第一性原理计算分析了由起始材料SiOx和Li2O形成的Li4SiO4和Li2SiO3等相。如图9(a)所示,Li2SiO3的形成能(−0.68 eV)小于Li4SiO4的形成能(−0.64 eV),说明在SiOx与Li2O的反应中,Li2SiO3更容易生成;Li2SiO3(2.529 g/mL)的密度高于Li4SiO4(2.42 g/mL)的密度,Li2SiO3的缓冲性能优于Li4SiO4,缓冲矩阵可以更好地缓解Si晶粒的体积膨胀。且Li2SiO3同样具有较好的锂离子传输的三维通道,为Li+嵌入/脱出硅颗粒提供有利的环境[30, 55]; Li2SiO3还具有比Li4SiO4更高的热稳定性,能够在电池循环过程中保持更加完整的结构,减少电池材料的衰减和损耗[56]。除此之外, Li2SiO3在水中的溶解度比Li4SiO4更小(微溶于水),与电池的制浆工艺的兼容性更高。从电子电导率看, Li2SiO3为1.72×10−9 S/cm,Li4SiO4为1.66×10−9 S/cm,Li2SiO3的高电导率可以提供更优异的倍率性能[57]。Li2SiO3相对水分和 CO2 气体的化学反应性而言也是最为稳定的[58],能够提高预锂化后的材料在空气中的稳定性。因此,在大多数的SiOx固相预锂化的反应中也主要以调控生成Li2SiO3相为主,以此带来更好的预锂化效果[30, 58-59]。
Yom等[60]使用锂金属与SiOx反应,探究了在不同加热温度及升温速率条件下对Li2SiO3与Li4SiO4生成的影响。发现在300~600℃的温度范围内,无论升温速率如何,Li2SiO3均在350℃左右开始形成(图9(b))。相反,Li4SiO4只能在450℃以上,升温速率在14℃/min或以上的条件下产生。Li2SiO3、Li4SiO4反应机制如下(<750℃)[56, 60-61] :
SiO2+4Li→Si+2Li2O (1) Li2O+SiO2→Li2SiO3 (2) Li2SiO3+Li2O→Li4SiO4 (3) 从时间-温度-变换(TTT)曲线亦可看出(图9(c)),在缓慢升温速率下有Li2SiO3的生成,但无法观察到Li4SiO4的形成。然而,在较高的升温速率下,有Li2SiO3和Li4SiO4的生成。Li2SiO3形成的温度比Li4SiO4更低,且在较低的升温速率下更容易调控Li2SiO3相的生成。此外,在SiOx固相预锂化中,过高的升温速率容易导致硅晶粒尺寸的快速增大以及加剧预锂化过程的反应不完全性以及预锂化不均匀性,而较低的升温速率能够带来更好的预锂化效果。因此,在缓慢升温速率下生成的Li2SiO3相更符合SiOx固相预锂化热处理工艺参数条件下的产物。
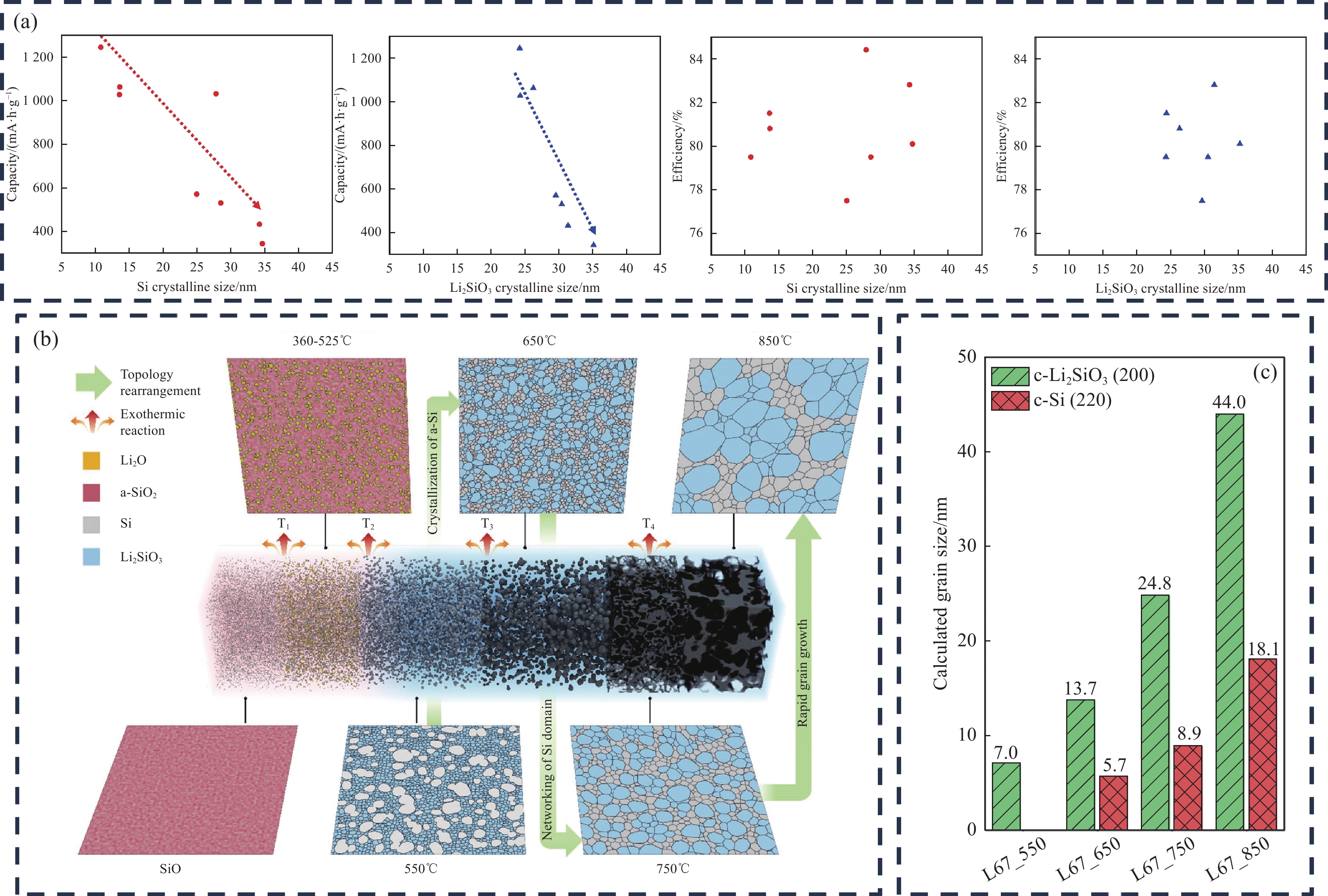
Bhat等[62]使用SLMP预锂化SiOx,探究了预锂化后Si和Li2SiO3的晶粒尺寸对电池容量与ICE的影响(图10(a))。当Si和Li2SiO3的晶粒尺寸增大时,呈现相似的趋势,容量随着尺寸的增加而减小。另一方面,ICE与Si和Li2SiO3的晶粒尺寸无关。Si和Li2SiO3的晶粒增大会使电子和锂离子的迁移路径更长,电阻和离子电阻会显著增大,会导致电池的循环稳定性变差。因此,在调控Li2SiO3相的生成需要注意其晶粒尺寸的变化,避免过大的晶粒对电池的性能造成影响。
![]() Figure 10. (a) Graph showing the relationship distribution between the grain size of Si and Li2SiO3 and their capacity, as well as ICE[62]; (b) Schematic illustration of phase transition and microstructure evolution driven by dehydrogenation-induced prelithiation[31]; (c) Dimensions of Si and Li2SiO3 at different temperatures[31]
Figure 10. (a) Graph showing the relationship distribution between the grain size of Si and Li2SiO3 and their capacity, as well as ICE[62]; (b) Schematic illustration of phase transition and microstructure evolution driven by dehydrogenation-induced prelithiation[31]; (c) Dimensions of Si and Li2SiO3 at different temperatures[31]Chung等[31]研究了LiH固相预锂化SiOx(Li/Si比为0.67)的拓扑排列结构,发现可通过不同的加热温度优化预锂化SiOx中Li2SiO3缓冲相与Si活性相的排列结构、降低晶粒尺寸,从而提升其电化学性能。图10(b)为不同温度下SiOx预锂化后不同相的相变和微观结构演化示意图。550~850℃呈现了不同温度下Li2SiO3缓冲相与Si活性相的拓扑排列结构。随着加热温度的升高,晶界不断发生迁移[63],相邻的Si、Li2SiO3晶粒彼此相互吞并,逐渐长大;拓扑结构不断发生改变,Si与Li2SiO3的相间排列性不断减弱。850℃时,Si与Li2SiO3的晶粒尺寸最大,分别为18.1 nm、44 nm (图10(c)),其预锂化后的循环稳定性最差,主要归咎于固相热处理中硅晶粒增大的问题[64],使得循环性能恶化[65]。且Si与Li2SiO3的相间排列性减弱,Li2SiO3对Si晶粒的体积膨胀的抑制效果降低。650℃时循环稳定性最好,Si与Li2SiO3的相间排列性均匀,晶粒尺寸相对较小(图10(b)),Li2SiO3对Si晶粒的体积膨胀也起到了一定的抑制效果。但650℃的热处理温度预锂化SiOx不完全,ICE(89.6%)相对750℃(92.7%)、850℃(91.2%)略有下降。
通过设置合理的热处理参数可以调控SiOx固相预锂化中Li2SiO3相的生成以及进一步优化Si活性相与Li2SiO3缓冲相的相间排列结构的均匀性。降低热处理温度以及升温速率可以避免晶粒的过度生长,从而提高固相预锂化后SiOx的循环稳定性。但在保证预锂化SiOx高ICE的前提,优化热处理参数,依旧难以避免硅晶粒在热处理过程中增大的趋势。因此,如何更大程度的优化SiOx固相预锂化的效果,仍然需要进一步的探索研究。
4. 固相预锂化的问题及解决方法
固相预锂化技术目前在产业化中主要存在3个问题:(1)固相热处理过程中硅晶粒尺寸的增大,降低其循环性能;(2)固相预锂化后,表面残锂、裸露的锂硅酸盐(Li2SiO3、Li2Si2O5、Li2SiO4)导致的强碱性环境,在材料电池制浆时明显的产气现象;(3)固相预锂化后,浆料的强碱性对粘结剂的粘结力变弱的影响[66-69]。
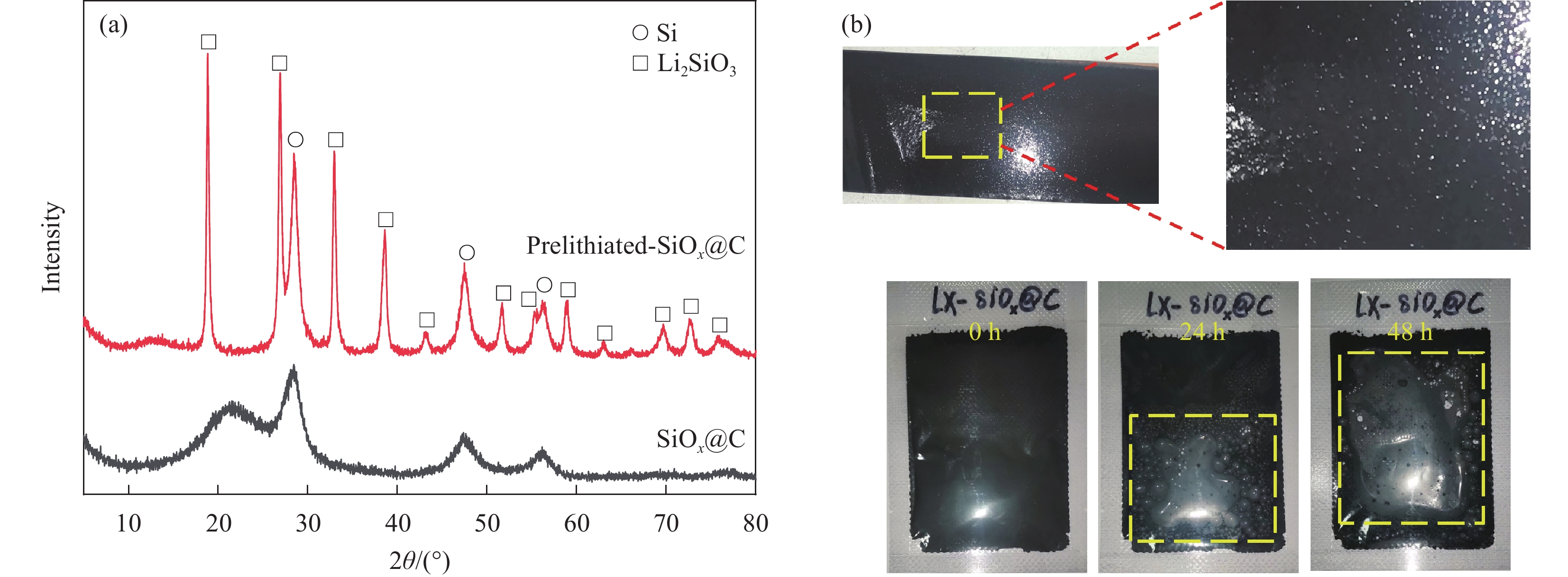
产生上述问题主要有以下原因:(1)硅晶粒尺寸的增大,主要由于在过高的热驱动力下晶界不断发生迁移,相邻的Si晶粒尺寸相互吞并,不断长大(表2、图11(a)为预锂化前后的硅晶粒尺寸);(2)浆料的产气现象是由于碱性溶液会与硅晶粒发生反应产生氢气[70-71],导致浆料产气现象。在全电池的制作过程中,SiOx较大的产气量,使得涂布完后极片会有较多的气孔产生,导致浆料与铜箔的粘着性差,难以使浆料较为均匀且紧密的分布在铜箔上(图11(b));(3)粘结剂的粘结力变弱是由于固相预锂化后SiOx表面残锂,以及裸露的锂硅酸盐(Li2SiO3、Li2Si2O5、Li2SiO4)微溶于水的性质,使得浆料pH值上升;而在负极材料的制浆工艺中,使用的大部分粘结剂受碱性的影响较大,如:聚丙烯酸(PAA)、羧甲基纤维素(CMC)、聚乙烯醇(PVA)、丁苯橡胶(SBR)和海藻酸盐(SA)等[72-74];如果浆料处于较大的pH值内(pH>11),高碱性浆料会在匀浆过程破坏粘结剂的基团,使得粘结力变弱;导致活性物质难以被粘结剂束缚于铜箔上,使电池在循环过程中发生脱落等现象,大大降低了电池的循环性能。
表 2 LiH预锂化SiOx@C前后硅晶粒尺寸对比Table 2. Comparison of silicon grain sizes before and after LiH prelithiation of SiOx@CScherrer formula (Dc) Calculated crystal face Silicon grain size/nm [(k×0.1λ/(cosθ×radians(FWHM))] (111) SiOx@C Prelithiated-SiOx@C 3.60 7.35 Notes: k—Scherrer constant (0.89); λ—X-ray wavelength (0.154 nm); θ—Bragg diffraction angle; FWHM—Half-width of the diffraction peak. ![]() 图 11 LiH预锂化SiOx前后XRD图谱(a)和全电池浆料产气评价(b)Figure 11. XRD patterns of SiOx before and after LiH prelithiation (a) and gas evolution evaluation of the full-cell slurry (b)
图 11 LiH预锂化SiOx前后XRD图谱(a)和全电池浆料产气评价(b)Figure 11. XRD patterns of SiOx before and after LiH prelithiation (a) and gas evolution evaluation of the full-cell slurry (b)针对上述问题,产业界主要通过优化热处理的工艺参数来减小硅晶粒尺寸,以及建立包覆层来解决浆料的产气、强碱性等问题。通过优化热处理的工艺参数,如:降低热处理温度、减小升温速率可在一定的程度上抑制热处理过程中硅晶粒尺寸的增长,但由于SiO2与锂化合物的反应需要在750℃左右才能反应完全[31],因此低于此热处理温度会导致预锂化不充分,残余一定量的SiO2,使得 ICE提升效果不足;且过低的升温速率也增加了工艺的生产成本。对预锂化后的SiOx进行多层包覆,可以降低浆料pH值、抑制浆料产气、保证涂布均匀。如:杜宁等[75]通过将预锂化后的SiOx置于含硅源的混合气氛中,经高温焙烧 后得到表面包覆硅的预锂化SiOx,再与有机碳源共混,高温煅烧进行了碳包覆处理,最后得到了由硅、碳双层包覆的预锂化SiOx。双层包覆后的预锂化SiOx的pH值可控制在10以下,解决了传统高碱性预锂化硅氧材料的产气问题、与粘结剂不适用等问题,同时极大地提高了首次库伦效率与电池容量。李波等[76]将预锂化后的SiOx先后进行了TiO2包覆、碳包覆。包覆后的预锂化SiOx可以将硅酸锂盐与水完全隔绝,使得材料即使长时间匀浆,其浆料的pH值也几乎没有变化,同样解决了高碱性的预锂化的材料带来的一系列问题。张健等[77]将预锂化后的SiOx先后包覆了聚合物层(含氟聚合物、聚苯乙烯和聚异戊二烯中的一种或多种)、碳层。使得内核中的锂硅酸盐被严密包覆,防止了水的渗入,解决了浆料pH升高、浆料产气等问题,保证了涂布的均匀性。虽然上述的多层包覆方法,可以较好地解决预锂化SiOx高碱性带来的一系列问题,但该项技术极大的增加了工艺的复杂度,提高了生产成本。
除上述研究之外,工业界也在开发与固相预锂化工艺相兼容的粘结剂,降低浆料的强碱性对其的影响;针对性的对热处理设备进行优化改造,使其具有散热功能好、密封性优良的特点,以降低热处理温度对硅晶粒长大的影响、提升预锂化效率等,以此提高固相预锂化方法的实用性。然而,实现高ICE的SiOx产业化生产仍需科研人员不断努力的探索。
5. 结语与展望
总体来看,固相预锂化由于其简单的生产工艺、对环境友好的特点以及对首次库仑效率(ICE)提升明显的效果,使其具备规模化生产的巨大潜力;其中用LiH作为固相预锂化锂源,干法混合热处理,对SiOx的ICE提升最为明显,循环寿命、倍率型方面的表现也十分优异。但固相预锂化需要在高温下才能促使锂源与SiO2发生反应,因此在热处理过程中容易使晶界发生迁移,硅晶粒尺寸增大,导致循环稳定性变差;预锂化后SiOx的残锂、裸露的锂硅酸盐,会导致浆料产气、pH值上升等问题,使其难以与制浆工艺完全兼容,对电池的生产造成了较大的阻碍。当前,主要通过降低热处理温度和建立包覆层等方法来解决硅晶粒尺寸增大、浆料产气和pH值升高等问题,但这会牺牲SiOx部分ICE并增加预锂化工艺的复杂度。对于固相预锂化热处理过程、浆料产气等反应机制仍然了解不足,需要进一步深入系统的研究。但随着研究的深入和技术的进步,固相预锂化的制备工艺流程将逐渐成熟完善,为锂离子电池的商业化发展做出更大贡献。
-
![]()
图 1 U-CF (a)、CF-5 (b)、CF-10 (c)、CF-20 (d)的SEM图像
Figure 1. SEM images of U-CF (a), CF-5 (b), CF-10 (c) and CF-20 (d)
![]()
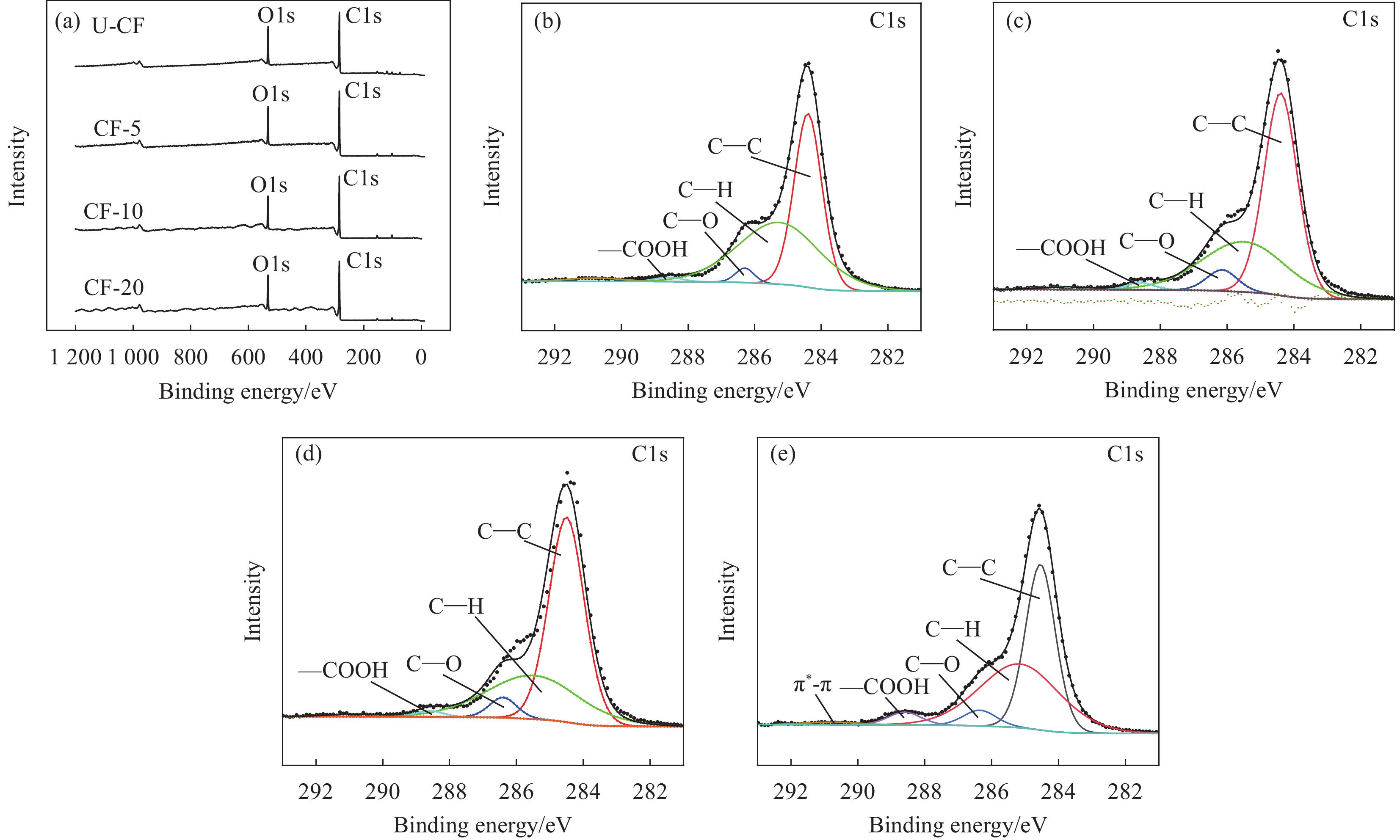
图 2 (a)高模量碳纤维(HMCF)的XPS广谱图;C1s的分峰拟合图谱:(b) U-CF;(c) CF-5;(d) CF-10;(e) CF-20
Figure 2. (a) XPS broad spectra of high-modulus carbon fiber (HMCF); C1s peak fitting diagrams: (b) U-CF; (c) CF-5; (d) CF-10; (e) CF-20
![]()
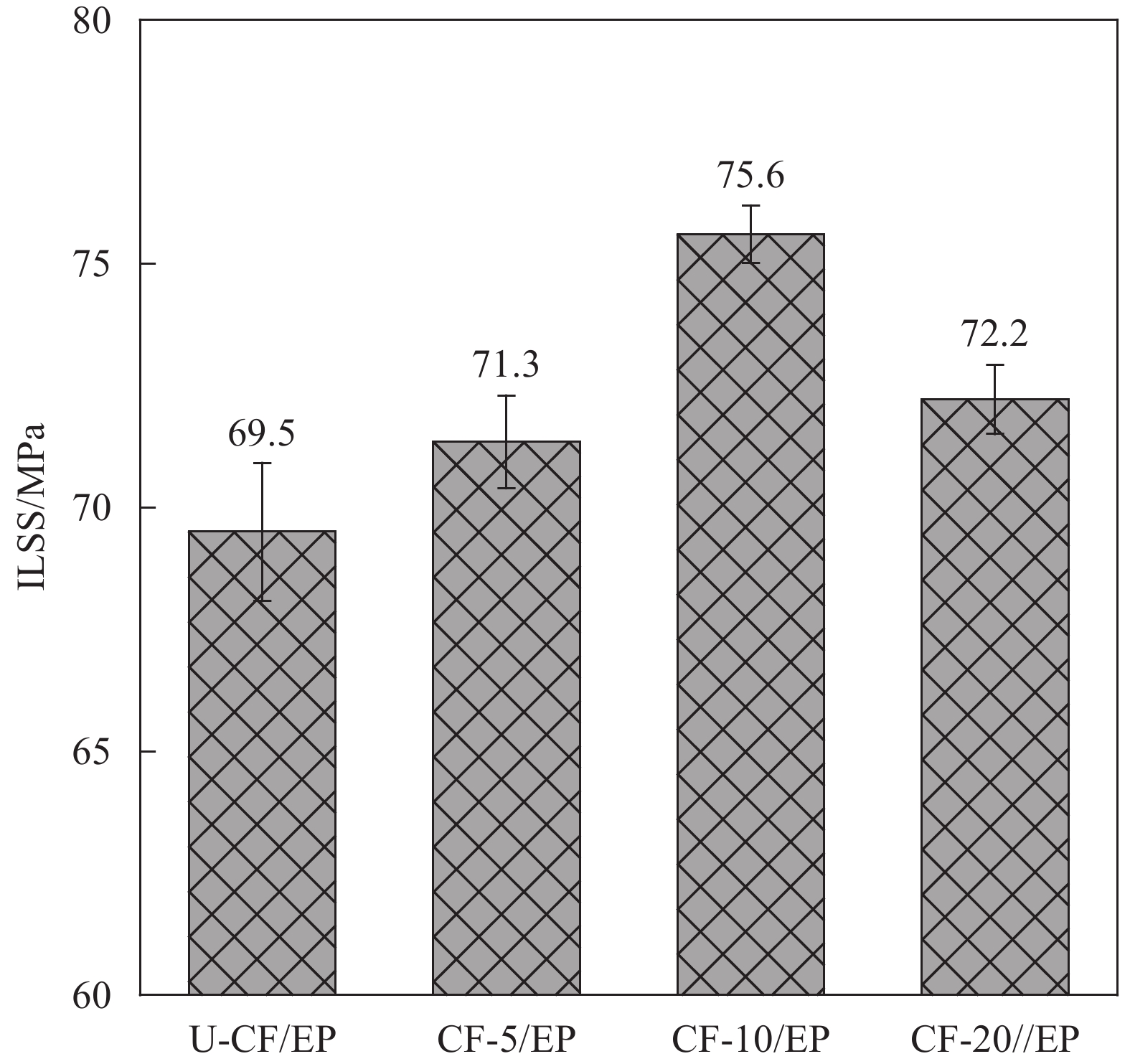
图 3 不同HMCF/EP复合材料的层间剪切强度(ILSS)
Figure 3. Interlaminar shear strength (ILSS) of different HMCF/EP composites
![]()
图 4 不同HMCF/EP复合材料横断面SEM图像:(a) U-CF/EP;(b) CF-5/EP;(c) CF-10/EP;(d) CF-20/EP
Figure 4. Cross-sectional SEM images of different HMCF/EP composites: (a) U-CF/EP; (b) CF-5/EP; (c) CF-10/EP; (d) CF-20/EP
![]()
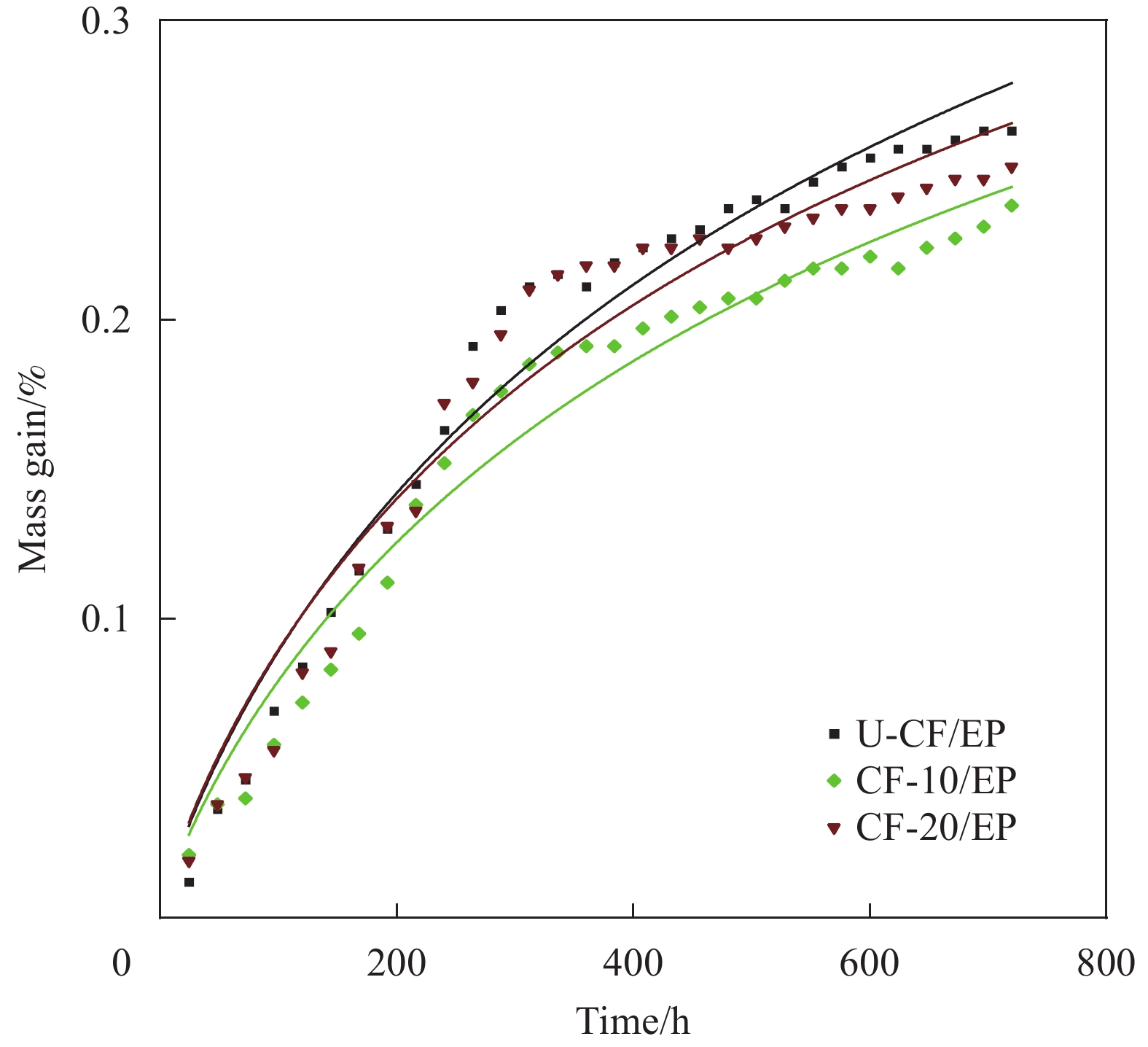
图 5 90℃、80% RH下HMCF/EP吸湿率随时间的Fick二段扩散模型拟合
Figure 5. Fitting of the Fick two-stage diffusion model of HMCF/EP moisture absorption over time at 90℃, 80% RH
![]()
图 6 CF-10/EP经不同湿热时间下复合材料的纵断面SEM图像:(a) 0 d;(b) 10 d;(c) 20 d;(d) 30 d
Figure 6. SEM images of profile of CF-10/EP under different hygrothermal time: (a) 0 d; (b) 10 d; (c) 20 d; (d) 30 d
![]()
图 7 不同HMCF/EP复合材料轴向截面形态SEM图像:(a) U-CF/EP;(a1) Aged U-CF/EP;(b) CF-10/EP;(b1) Aged CF-10/EP;(c) CF-20/EP;(c1) Aged CF-20/EP
Figure 7. SEM images of axial cross-section morphology of different HMCF/EP composites: (a) U-CF/EP; (a1) Aged U-CF/EP; (b) CF-10/EP; (b1) Aged CF-10/EP; (c) CF-20/EP; (c1) Aged CF-20/EP
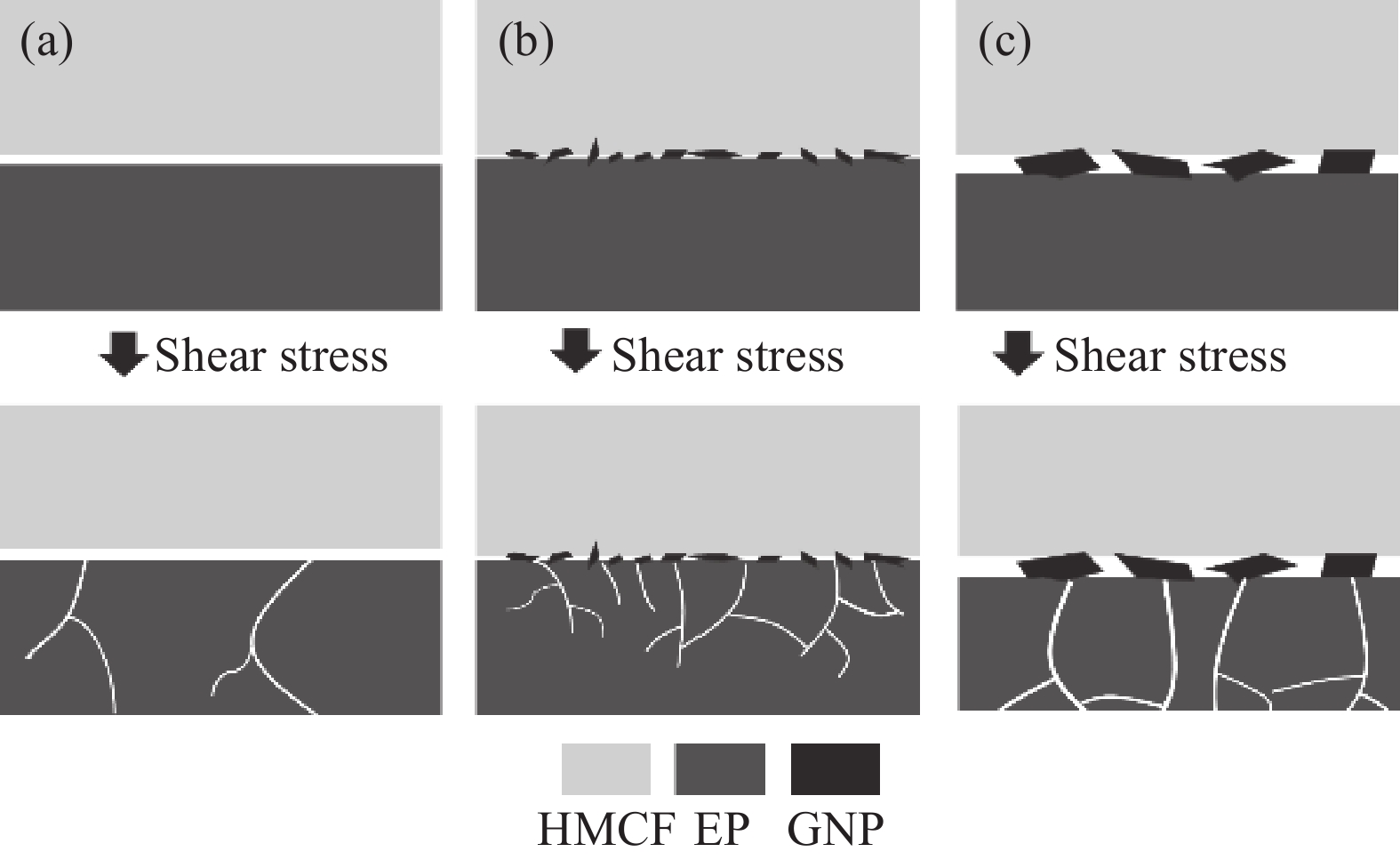
![]()
图 8 不同HMCF/EP复合材料经湿热试验后的界面失效机制:(a) U-CF/EP;(b) CF-10/EP;(c) CF-20/EP
Figure 8. Interfacial failure mechanisms of different HMCF/EP composites after hygrothermal test: (a) U-CF/EP; (b) CF-10/EP; (c) CF-20/EP
GNP—Graphene nanosheets
表 1 各样品缩写命名
Table 1 Abbreviations used for various samples prepared
Sample code Details U-CF Untreated commercial carbon fibers (CF) CF-5 0.5 mg/mL graphene nanoplates solution 5 V voltage electrophoresis deposited modified fibers CF-10 0.5 mg/mL graphene nanoplates solution 10 V voltage electrophoresis deposited modified fibers CF-20 0.5 mg/mL graphene nanoplates solution 20 V voltage electrophoresis deposited modified fibers U-CF/EP Untreated commercial carbon fiber reinforced epoxy (EP) composites CF-5/EP 0.5 mg/mL graphene nanoplates solution 5 V voltage electrophoresis deposited modified fibers reinforced epoxy composites CF-10/EP 0.5 mg/mL graphene nanoplates solution 10 V voltage electrophoresis deposited modified fibers reinforced epoxy composites CF-20/EP 0.5 mg/mL graphene nanoplates solution 20 V voltage electrophoresis deposited modified fibers reinforced epoxy composites Aged U-CF/EP After 30 d of hygrothermal test (90℃, 80% relative humidity (RH)) untreated commercial carbon fibers reinforced epoxy composites Aged CF-5/EP After 30 d of hygrothermal test (90℃, 80% RH) 0.5 mg/mL graphene nanoplates solution 5 V voltage electrophoresis deposited modified fibers reinforced epoxy composites Aged CF-10/EP After 30 d of hygrothermal test (90℃, 80% RH) 0.5 mg/mL graphene nanoplates solution 10 V voltage electrophoresis deposited modified fibers reinforced epoxy composites Aged CF-20/EP After 30 d of hygrothermal test (90℃, 80% RH) 0.5 mg/mL graphene nanoplates solution 20 V voltage electrophoresis deposited modified fibers reinforced epoxy composites  下载: 导出CSV
下载: 导出CSV
表 2 不同HMCF C1s特征峰结合能及相对含量
Table 2 C1s characteristic peak binding energy and relative content of different HMCF
Functional group Binding energy/eV U-CF/wt% CF-5/wt% CF-10/wt% CF-20/wt% C—C 284.4 46.60 48.31 49.36 44.56 C—H 285.2 48.78 46.08 46.59 45.76 C—O 286.3 2.19 2.69 1.35 4.65 C=O 287.3 — — — — COOH 288.6 0.72 1.70 3.79 3.78
下载: 导出CSV
表 3 90℃、80% RH湿热条件下HMCF/EP的吸湿率
Table 3 Moisture absorption of HMCF/EP at 90℃, 80% RH
Sample M5 d/% M10 d/% M15 d/% M20 d/% M25 d/% M30 d/% U-CF 0.08 0.16 0.21 0.24 0.25 0.26 CF-10/EP 0.07 0.15 0.19 0.21 0.22 0.24 CF-20/EP 0.08 0.17 0.22 0.22 0.24 0.25 Note: Mx d—Moisture absorption of composite material after x days.
下载: 导出CSV
表 4 80% RH、90℃湿热老化HMCF/EP吸湿拟合参数
Table 4 Hygroscopic fitting parameters of HMCF/EP for hygrothermal at 90℃, 80% RH
D/
(10−4 mm·h−1)M1∞/% M30 d/% k/
(10−2 mm·h−1/2)R2 CF-10/EP 14.82 0.12 0.24 4.43 0.96 CF-20/EP 15.13 0.14 0.25 3.89 0.95 U-CF/EP 15.69 0.12 0.26 5.19 0.97 Notes: D—Diffusion coefficient of water molecules in composites; M1∞—Saturated moisture absorption rate of the first stage of the composite Fick; M30 d—Moisture absorption of composite material after 30 d; k—Constant related to the relaxation of resin structure; R2—Coefficient of determination.
下载: 导出CSV
表 5 80% RH、90℃下HMCF/EP老化过程中层间剪切强度
Table 5 Interlaminar shear strength during HMCF/EP aging at 90℃, 80% RH
Sample ILSS0 d/
MPaILSS10 d/
MPaILSS20 d/
MPaILSS30 d/
MPaILSS60 d/
MPaU-CF/EP 69.5 68.6 65.4 64.4 61.3 CF-10/EP 75.6 73.2 69.3 69.5 67.8 CF-20/EP 72.2 68.9 68.1 66.7 65.5 Note: ILSSx d—Interlaminar shear strength of composite material after x days.
下载: 导出CSV
-
[1] 陶积柏, 黎昱, 张玉生, 等. 高模量碳纤维在中国宇航结构产品上的应用现状及实现自我保障的建议[J]. 材料科学与工艺, 2015, 23(6): 98-103. DOI: 10.11951/j.issn.1005-0299.20150618 TAO Jibai, LI Yu, ZHANG Yusheng, et al. Application status of high-modulus carbon fiber in domestic aerospace structural products and suggestions for self-supply[J]. Materials Science and Technology, 2015, 23(6): 98-103(in Chinese). DOI: 10.11951/j.issn.1005-0299.20150618
[2] BARAL N, DAVIES P, BALEY C, et al. Delamination behaviour of very high modulus carbon/epoxy marine composites[J]. Composites Science and Technology, 2008, 68(3-4): 995-1007. DOI: 10.1016/j.compscitech.2007.07.022
[3] HAO R, JIAO X, ZHANG X, et al. Fe3O4/graphene modified waterborne polyimide sizing agent for high modulus carbon fiber[J]. Applied Surface Science, 2019, 485: 304-313. DOI: 10.1016/j.apsusc.2019.04.184
[4] LIN J, XU P, WANG L, et al. Multi-scale interphase construction of self-assembly naphthalenediimide/multi-wall carbon nanotube and enhanced interfacial properties of high-modulus carbon fiber composites[J]. Composites Science and Technology, 2019, 184(10): 107855.
[5] GAO S L, MADER E, ZHANDAROV S F, et al. Carbon fibers and composites with epoxy resins: Topography, fractography and interphases[J]. Carbon, 2004, 42(3): 515-529. DOI: 10.1016/j.carbon.2003.12.085
[6] MCCRARY D M C, OKOLI O I. A review of multiscale composite manufacturing and challenges[J]. Journal of Reinforced Plastics and Composites, 2012, 31(24): 1687-1711. DOI: 10.1177/0731684412456612
[7] QI X, TIAN J, XIAN G. Hydrothermal ageing of carbon fiber reinforced polymer composites applied for construction: A review[J]. Journal of Materials Research and Technology, 2023, 27: 1017-1045. DOI: 10.1016/j.jmrt.2023.09.198
[8] SUN P, ZHAO Y, LUO Y. Effect of temperature and cyclic hygrothermal aging on the interlaminar shear strength of carbon fiber/bismaleimide (BMI) composite[J]. Materials and Design, 2011, 32(8-9): 4341-4347. DOI: 10.1016/j.matdes.2011.04.007
[9] DZUL-CERVANTES M A A, PACHECO-SALAZAR O F, CAN-HERRERA L A, et al. Effect of moisture content and carbon fiber surface treatments on the interfacial shear strength of a thermoplastic-modified epoxy resin composites[J]. Journal of Materials Research and Technology, 2020, 9(6): 15739-15749.
[10] YU B, JIANG Z, YANG J. Long-term moisture effects on the interfacial shear strength between surface treated carbon fiber and epoxy matrix[J]. Composites Part A: Applied Science and Manufacturing, 2015, 78: 311-317.
[11] ZHANG S, LIU W B, HAO L F, et al. Preparation of carbon nanotube/carbon fiber hybrid fiber by combining electrophoretic deposition and sizing process for enhancing interfacial strength in carbon fiber composites[J]. Composites Science and Technology, 2013, 88: 120-125. DOI: 10.1016/j.compscitech.2013.08.035
[12] WU G, MA L, LIU L, et al. Interface enhancement of carbon fiber reinforced methylphenylsilicone resin composites modified with silanized carbon nanotubes[J]. Materials and Design, 2016, 89: 1343-1349.
[13] ASADI A, MILLER M, MOON R J, et al. Improving the interfacial and mechanical properties of short glass fiber/epoxy composites by coating the glass fibers with cellulose nanocrystals[J]. Abstracts of Papers of the American Chemical Society, 2016, 10(7): 587-597.
[14] MOON R J, MARTINI A, NAIRN J, et al. Cellulose nanomaterials review: Structure, properties and nanocomposites[J]. Chemical Society Reviews, 2011, 40(7): 3941-3994. DOI: 10.1039/c0cs00108b
[15] 刘文军, 严建龙, 周川, 等. 氧化石墨烯改性碳纤维/环氧树脂复合材料的湿热性能及微观形貌[J]. 复合材料学报, 2021, 38(5): 1416-1425. LIU Wenjun, YAN Jianlong, ZHOU Chuan, et al. Hygrothermal properties and micro morphology of graphene oxide modified carbon fiber/epoxy resin composites[J]. Acta Materiae Compositae Sinica, 2021, 38(5): 1416-1425(in Chinese).
[16] ZHUANG X, MA J, DAN Y, et al. Hydrothermal aging of carbon fiber reinforced epoxy composites with different interface structures[J]. Polymer Degradation and Stability, 2023, 212: 110352. DOI: 10.1016/j.polymdegradstab.2023.110352
[17] KONG N, KHALIL N Z. Water absorption mechanism and mechanical performance of adhesively joint aluminum alloy with GNP reinforced epoxy[J]. Materials Today, 2022, 51(2): 1392-1398.
[18] KONG N U, KHALIL N Z, FRICKE H. Moisture absorption behavior and adhesion properties of GNP/epoxy nanocomposite adhesives[J]. Polymers, 2021, 13(11): 1850. DOI: 10.3390/polym13111850
[19] OZGE K, LISA M, ASADI A. Cellulose nanocrystal-enabled tailoring of the interface in carbon nanotube and graphene nanoplatelet-carbon fiber polymer composites: Implications for structural applications[J]. ACS Applied Nano Materials, 2022, 5(1): 1284-1295. DOI: 10.1021/acsanm.1c03860
[20] 中国国家标准化管理委员会. 聚合物基复合材料短梁剪切强度试验方法: GB/T 30969—2014[S]. 北京: 中国标准出版社, 2014. Standardization Administration of the People's Republic of China. Test method for shear strength of short beams of polymer matrix composites: GB/T 30969—2014[S]. Beijing: Standards Press of China, 2014(in Chinese).
[21] QIAN X, ZHANG Y G, WANG X F, et al. Effect of carbon fiber surface functionality on the moisture absorption behavior of carbon fiber/epoxy resin composites[J]. Surface and Interface Analysis, 2016, 48(12): 1271-1277. DOI: 10.1002/sia.6031
[22] ZHONG Y, CHENG M, ZHANG X, et al. Hygrothermal durability of glass and carbon fiber reinforced composites—A comparative study[J]. Composite Structures, 2019, 211: 134-143. DOI: 10.1016/j.compstruct.2018.12.034
[23] BAO L R, YEE A F. Mositure diffusion and hygrothermal aging bismaleimide matrix carbon fiber composites: Part II—Woven and hybrid composites[J]. Composites Science and Technology, 2002, 62(16): 2111-2119.
[24] TSOTSIS T K, WEITSMAN Y. A simple graphical method for determining diffusion parameters for two-stage sorption in composites[J]. Journal of Materials Science Letters, 1994, 13: 1635-1636.
[25] VUKOVIC F, WALSH T R. Moisture ingress at the molecular scale in hygrothermal aging of fiber-epoxy interfaces[J]. ACS Applied Materials and Interfaces, 2020, 12(49): 55278-55289. DOI: 10.1021/acsami.0c17027
-
其他相关附件
-
-
高模碳纤维以其密度低、模量高、热稳定性高等优点,在航天领域应用较多。但由于其增强复合材料的界面性能较弱影响其应用。
本文采用电泳沉积法将经纤维素纳米晶(CNC)分散的石墨烯纳米片(GNP)负载到高模碳纤维(HMCF)的表面,通过改变电压来调控HMCF表面特性,进而改善HMCF与环氧树脂(EP)的相互作用,使得HMCF复合材料的层间剪切强度达到75.6 MPa,提升了8.8%。与此同时,利用GNP的片层状特性在界面产生良好的阻隔效应,降低了复合材料的吸湿率:在90℃、80RH%环境下30天的吸湿率从未改性的0.263%降低至0.238%。在此湿热条件下存放60天后,复合材料的层间剪切仍保持在67.8 MPa,表现出较好的耐湿热性能。
HMCF增强树脂基复合材料层间剪切强度(a)和吸湿率(b)





计量
- 文章访问数: 179
- HTML全文浏览量: 87
- PDF下载量: 19
